Enhancing Efficiency: Fast and Reliable Nitrite Content Analysis in Pharmaceutical Excipients With the ACQUITY™ QDa™ II Mass Detector
Abstract
Benefits
- Demonstrate the robust capabilities of the ACQUITY™ QDa™ II Mass Detector through a comprehensive 29-day analysis, showcasing its use for routine determination of nitrite content in typical pharmaceutical excipients
- Highlight ease of use of the instrument, providing practical insights to operators regarding maintenance intervals, documenting any instrument downtime to evaluate suitability for high-sample throughput laboratories
- Assess method precision in quantifying nitrites at µg/g levels within the context of routine analytical testing, evaluating system reliability in applied workflows
- Employ Empower™ Chromatography Data System (CDS) software to ensure complaint and comprehensive reporting of results
Introduction
The presence of N-Nitrosamines in pharmaceutical products has been a rapidly evolving concern within the pharmaceutical industry in recent years. Global regulatory authorities have raised safety concerns regarding N-Nitrosamine contamination in several drug classes, owing to the idea that long-term exposure to such impurities, above a level that is considered safe, may confer carcinogenic and mutagenic risks for patients.1,2 Consequently, mitigating risk and controlling the presence of these compounds has become an integral part of pharmaceutical product development and quality evaluation.
Nitrite is the most critical risk factor in N-Nitrosamine formation.3 Reacting with secondary or tertiary amines, typically under acidic conditions, nitrite can form a reactive NO+ carrier species, which can act as a nitrosating agent. The indiscriminate interaction of this nitrosating agent with vulnerable amines under certain conditions, will significantly contribute to the level of all possible N-Nitrosamines present within a final pharmaceutical product. As a result, screening for total nitrite content as a single underlying precursor to such formation can be a simple and pragmatic approach to support risk assessment, which can efficiently mitigate the risk of N-Nitrosamine formation. This will help to reduce the requirement to perform traditional nitrosamine analyses, which are typically more challenging due to the sensitivity requirements.
Nitrite contamination is typically introduced from raw materials, intermediates, and excipients, as well as solvents and reagents during synthesis. However, nitrite present as an impurity in excipients is typically found in parts per million (ppm) and is the primary source of nitrosating agents.4,5 Variability in nitrite levels is well recognised across excipient types, but also across excipient suppliers. The same excipient purchased across different vendors can show starkly different levels of nitrite concentration, perhaps owing to differences in the manufacturing process.6
The wider variety of sample matrices and increased sample testing volume of this approach underlines the need for a routine method to assess nitrite concentration in excipients, allowing pharmaceutical manufacturers to have confidence that raw materials used are sufficiently free from potential contamination - irrespective of supplier, excipient type, or even batch used. Such a method should allow for flagging of contaminated raw materials, which can then be redirected for further confirmatory testing if deemed necessary. This risk assessment should be rapid and cost-effective, and while low-cost methods are commonly used in nitrite evaluation, they are often time-consuming and suffer from poor sensitivity and interferences.7
Liquid chromatography coupled to single quadrupole mass spectrometry (LC-MS) offers the sensitivity and selectivity needed for trace level analysis of nitrite, but ultimately any solution must also demonstrate robustness and reliability required for high-throughput routine workflows, while maintaining a low cost of analysis per sample.8
In the experiments described below, a UHPLC coupled to single quadrupole mass detector was employed to assess the suitability of this analytical configuration for long term analysis of pharmaceutical excipients over the course of 29 days of analysis. The objective of this work was to demonstrate the reliability and robust performance of the ACQUITY QDa II Mass Detector over time. The advantages of the proposed method, adopted from a UPLC™ method by Jireš & Douša, are speed of analysis, sensitivity, and selectivity of detection.8 For this analysis, an existing method was transferred to an ACQUITY Arc™ System, a cost-effective alternative which still demonstrates the performance, robustness, and reliability required for the effective analysis of pharmaceutical excipients.
Experimental
Commercially available excipient samples were purchased and kept at room temperature until use. Sorbitol, Lactose Monohydrate, Maize Starch, Maltodextrin, and Calcium Carbonate were selected to be representative of typical pharmaceutical excipients. All chemicals and reagents purchased were of analytical grade quality, and UPLC grade solvents were used for the preparation of all samples, solutions, and mobile phases.
Samples were extracted following a modified protocol from Jireš & Douša shown in Figure 1, where powdered excipients were taken through a 2,3-diaminonaphthalene (DAN) derivatisation method.8 The principle of this method is the reaction of nitrite ions with DAN to form 1H-Naphtho(2,3-d)triazole (NAT), which can then be separated and detected using mass detection. The NAT compound is then quantified as a proxy for total nitrite formation in the sample.
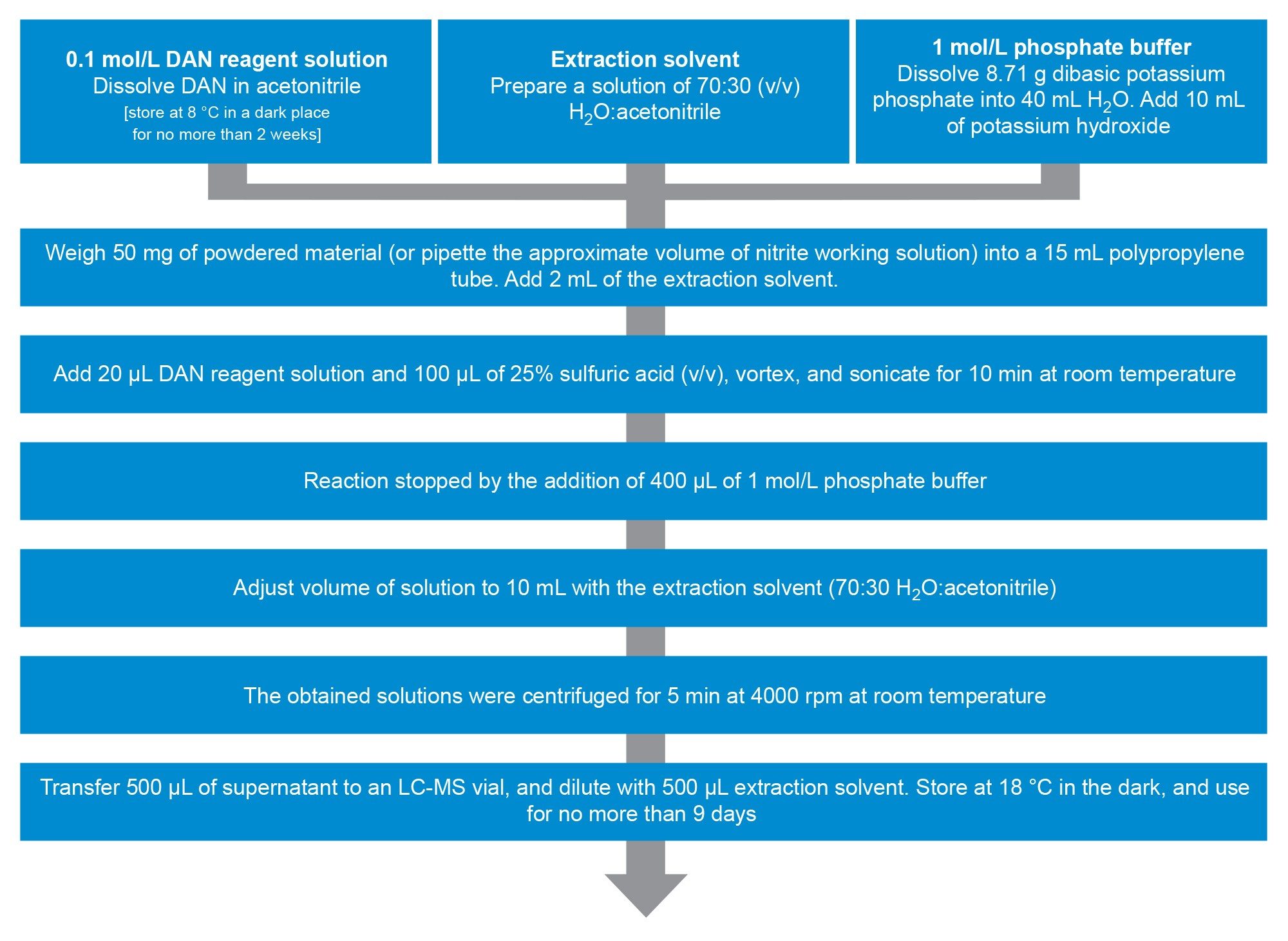 Figure 1. Sample preparation method adopted from Jireš & Douša, 2022.
Figure 1. Sample preparation method adopted from Jireš & Douša, 2022.
Construction of the calibration curve was performed using a nitrite spiking solution (1 µg/mL), which was added to empty polypropylene tubes at various concentrations (0.5, 5.0, and 12.5 ng/mL, corresponding to 0.2, 2.0, and 5.0 µg/g in excipient sample) and accordingly taken through the sample preparation procedure. Unspiked excipient samples were then quantified against the calibration curve. A process blank (procedural blank) sample was also prepared to assess any baseline level of nitrite interaction inherent in the sample preparation procedure. 100 µL of extraction solvent was used in place of a spike in the process blank.
Recoveries were assessed by spiking the corresponding amount of solution onto 50 mg of each excipient type at each spiking level (0.5, 5.0, and 12.5 ng/mL). All sample preparation for each batch was performed in triplicate.
To assess and monitor system stability for the purposes of the robustness test, instrument QC standards containing acetaminophen and caffeine were prepared in extraction solvent at two concentration levels (0.5 and 5 µg/mL) and were analysed throughout the course of the analysis.
Samples were prepared using the described sample preparation technique over a period of four sequential sample batches, meaning that the entire sample preparation procedure was repeated a total of four times. A selected ion recording (SIR) was acquired using a target ion at m/z 170.1 as [M+H]+ for NAT, m/z 152.1 [M+H]+ for acetaminophen, and m/z 195.1 [M+H]+ for caffeine.
Analysis was performed using an ACQUITY Arc System coupled to an ACQUITY QDa II Mass Detector. The Columns Calculator 2.0 Software (667005222) was used to ensure that UPLC conditions specified in a method by Jireš & Douša could be adopted and transferred to a UHPLC system while remaining within the pressure limits of the system.8 This allowed for an LC system with decreased specifications to be used without compromising expected performance and sensitivity of the overall system, ensuring cost-effective analysis.
LC-MS Experimental Conditions
|
LC system: |
ACQUITY Arc with FTN-R Sample Manager |
|
Detection: |
ACQUITY QDa II Mass Detector |
|
Column(s): |
ACQUITY UPLC HSS™ T3 Column, 100 Å, 1.8 µm, 2.1 mm x 100 mm (p/n: 186003539) |
|
Column temperature: |
45 °C |
|
Sample temperature: |
18 °C |
|
Injection volume: |
5 μL |
|
Flow rate: |
0.4 mL/min |
|
Run time: |
6 mins |
|
Mobile phase A: |
0.1% Formic Acid in H2O |
|
Mobile phase B: |
Acetonitrile |
|
Vials: |
Clear Glass 12 x 32 mm Screw Neck Vial, 100/pk (p/n: 186000273) |
|
Ionization: |
Positive Electrospray (ES+) |
|
Capillary voltage: |
0.8 kV |
|
Desolvation temperature: |
400 °C |
|
Source temperature: |
120 °C |
|
Acquisition mode: |
SIR |
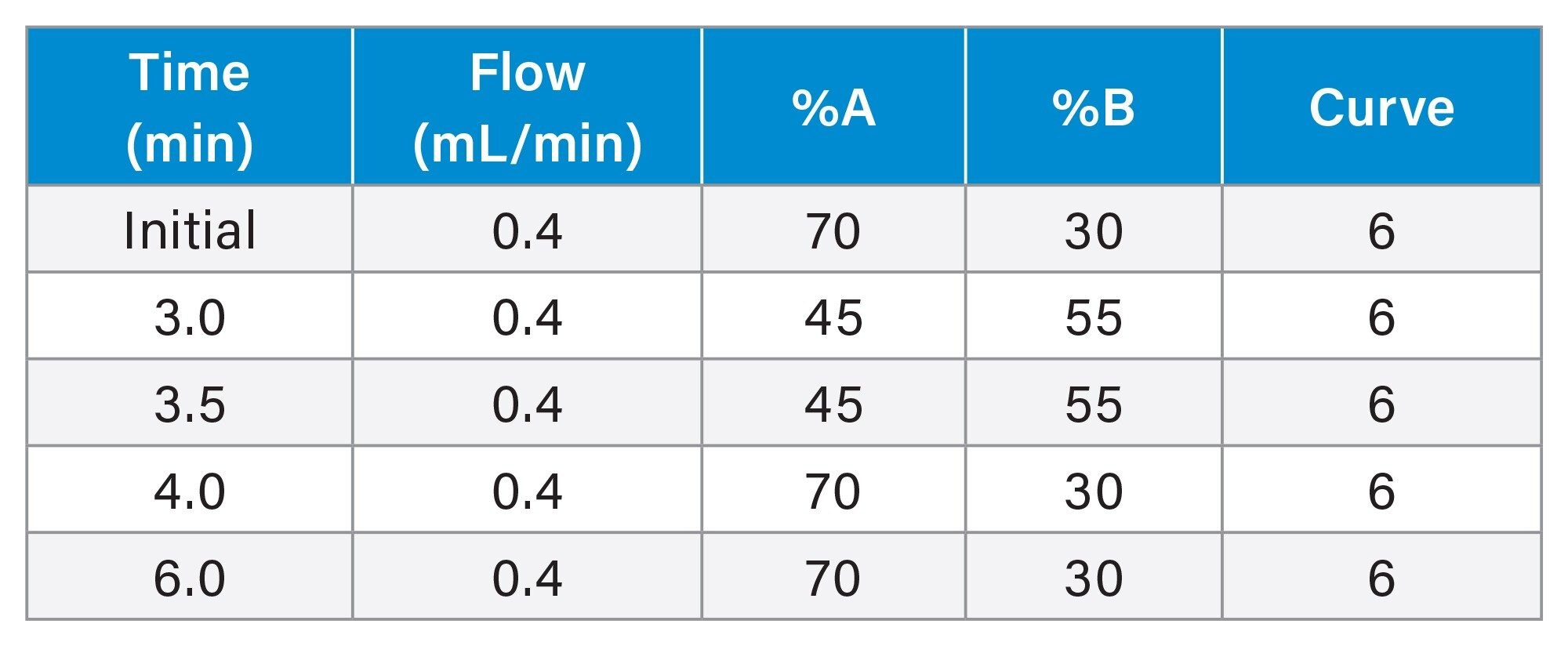
LC Gradient
Software
|
Data acquisition, processing, and reporting: |
Empower™ CDS 3.0 |
Results and Discussion
Method Performance
Using the LC-MS method conditions described above, fast determination of nitrite content in selected excipients was achieved by quantifying NAT (m/z 170). The NAT compound had a retention time (RT) of ~2.73 mins. Samples were quantified using a calibration curve ranging from 0.5–12.5 ng/mL, corresponding to 0.2–5.0 µg/g of nitrite concentration in the excipient samples (µg/g = ppm).
The LOQ of the method is limited by the presence of nitrite in the process blank. An example of this is illustrated in Figure 2 where absolute peak area of nitrite standards are shown alongside a process blank.
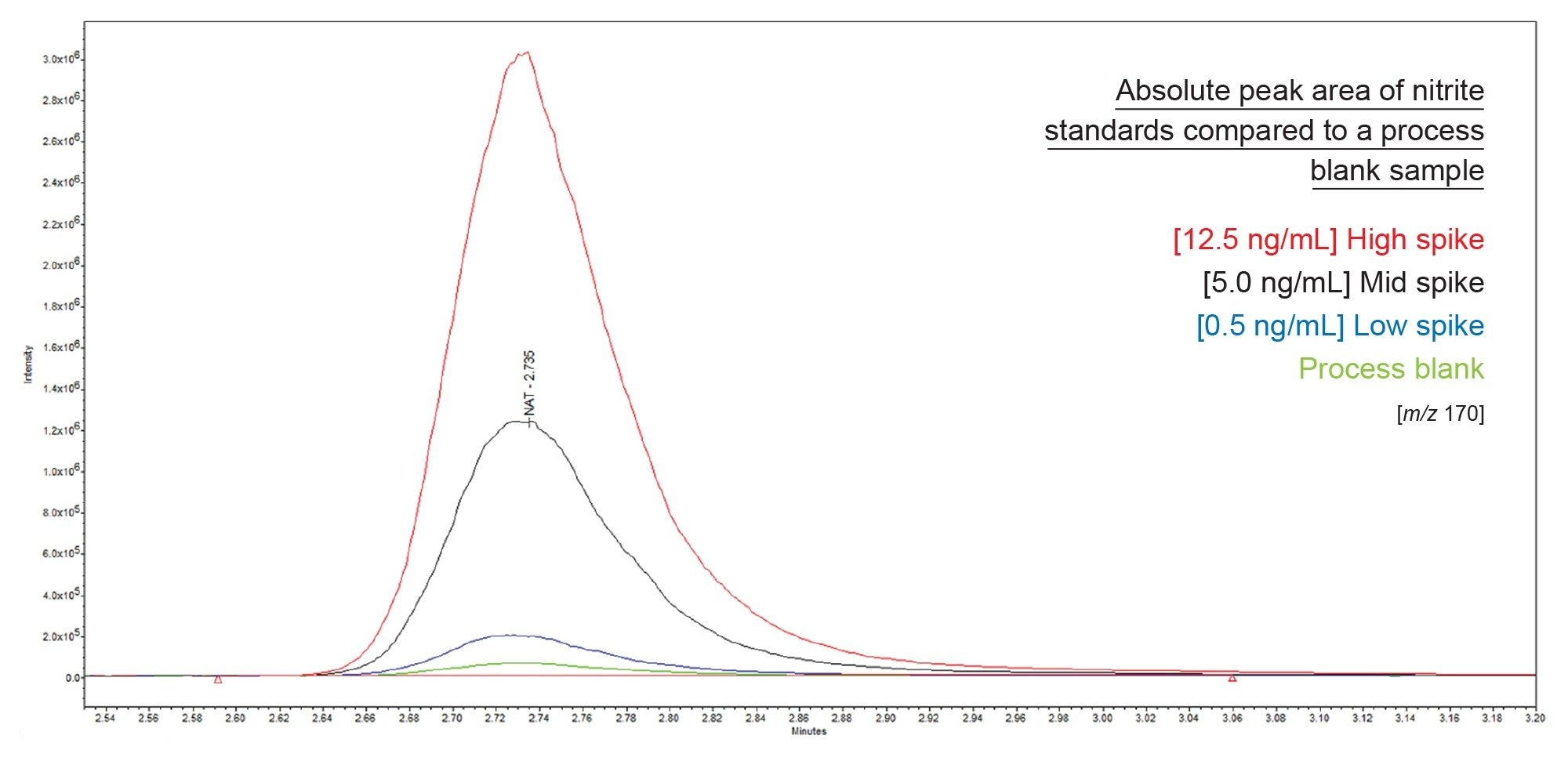 Figure 2. Absolute peak area of nitrite standards overlayed against a process blank for a given replicate of a sample sequence.
Figure 2. Absolute peak area of nitrite standards overlayed against a process blank for a given replicate of a sample sequence.
Calibration plots were constructed using Corrected Area, where an average value for the peak area of the process blank (n=6 in each sample set) was summed and then subtracted from the peak area of all standards and unknowns using the following formula: Corrected Area = Area – (BL?.%..AVE(Area)). Using this method of blank subtraction, the X-intercept of the adjusted calibration plot becomes a representative value for the process blank - the level of nitrite present in each sample due to contamination in the sample preparation technique. The increase in response above the intercept is then used to quantify the nitrite content in subsequent samples.
A predefined injection sequence (181 injections), intended to be a representation of a high throughput workflow, was created and analysed multiple times over the course of a 148–190-hour sample batch. Each sample batch here (n=4) denotes the preparation of new samples - including spiked and unspiked excipients, nitrite standards, process blanks, and QCs. Sample preparation was repeated four times over a total of 29 days of analysis, totalling 32 replicates of the injection sequence. The analysis was designed to demonstrate the robustness of the system, and was intended to be continuous and without interruption, other than for routine replacement of LC mobile phase and solvents. Any additional interaction, such as source cleaning, was recorded in Table 1.
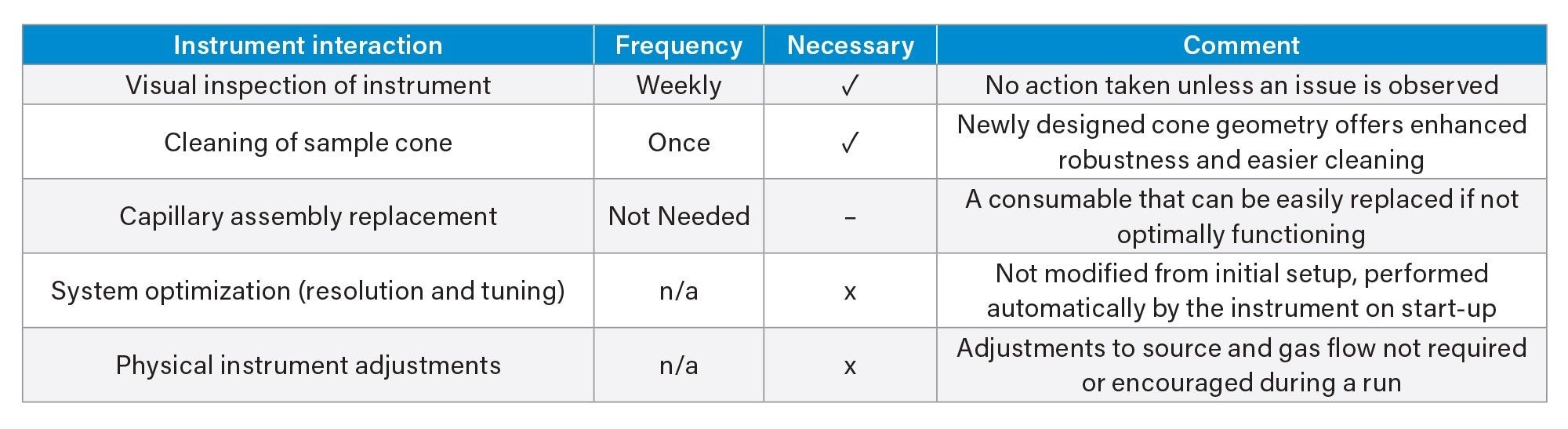 Table 1. Typical instrument interaction with the ACQUITY QDa II Mass Detector, and frequency of each task performed over the duration of the analysis.
Table 1. Typical instrument interaction with the ACQUITY QDa II Mass Detector, and frequency of each task performed over the duration of the analysis.
A calibration curve was plotted in Empower for each replicate of the predefined injection sequence (n=32) across the analysis. Each injection sequence was processed individually, and contained excipient samples bracketed by calibration standards, all of which were prepared in triplicate. The calibration curve for each injection sequence was linear (1/X) with an R2 >0.99 in all cases, apart from in sample batch 2 (R2 >0.97). Shown in Figure 3 is a comparison of the first and last calibration plots of a given sample batch, separated by almost 150 hours of analysis. In this example, the corresponding residual value for any given datapoint does not exceed 3.7%.
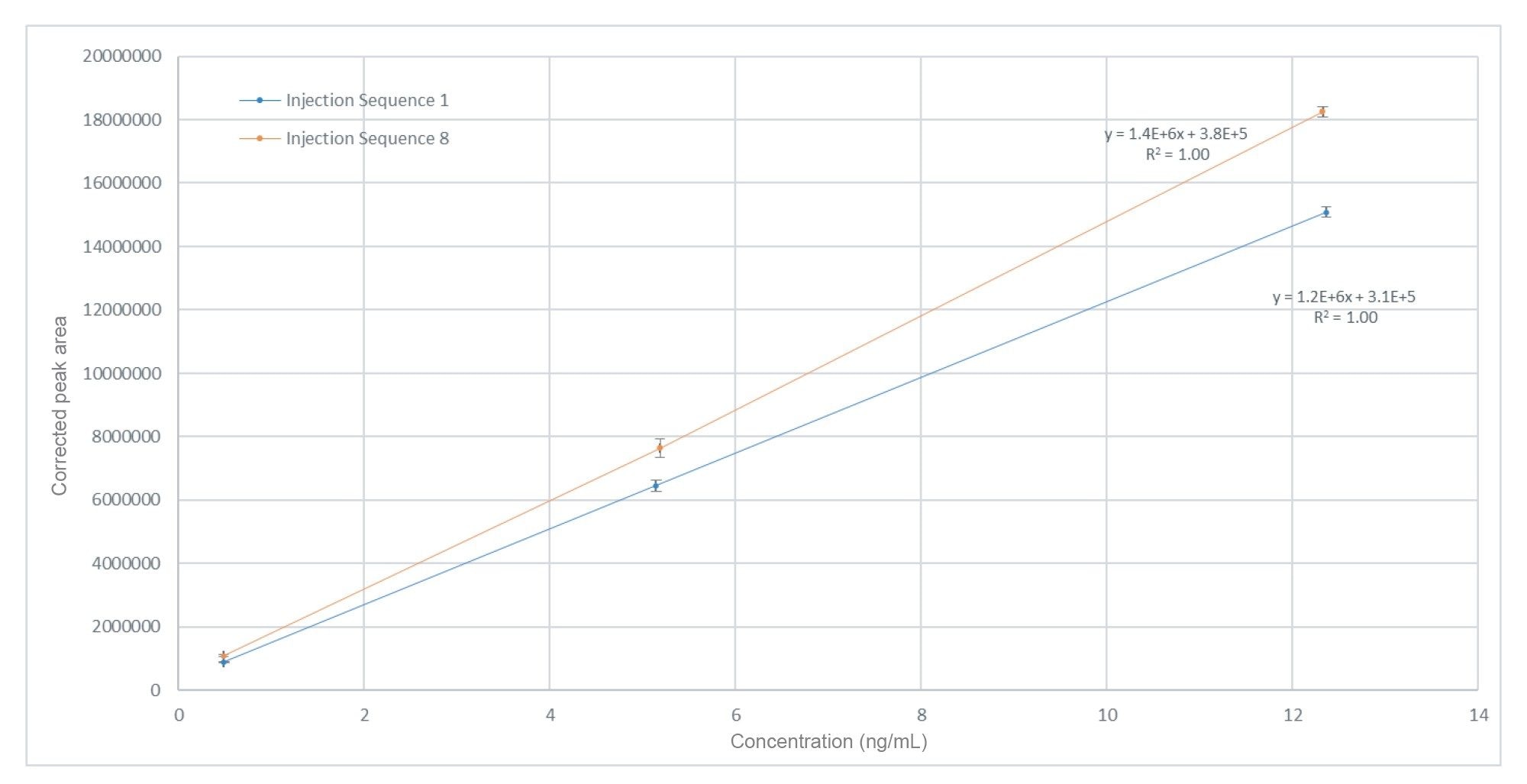 Figure 3. Nitrite (as NAT) calibration responses over a concentration range of 0.5–12.5 ng/mL (equivalent to 0.2–5 µg/g of nitrite in the raw material), analysed in triplicate injections for calibration standards at the beginning and end of a sample batch. Average values of each calibration level with respective standard deviation over n=6 injections.
Figure 3. Nitrite (as NAT) calibration responses over a concentration range of 0.5–12.5 ng/mL (equivalent to 0.2–5 µg/g of nitrite in the raw material), analysed in triplicate injections for calibration standards at the beginning and end of a sample batch. Average values of each calibration level with respective standard deviation over n=6 injections.
The final sample batch was analysed over a slightly extended period (190 hours) compared to the previous sample batches and is therefore best placed to show the stability of the method over time. For the purpose of this work, recoveries were assessed by comparing the peak area of the spiked excipients to that of a known equivalent spike level. Recoveries across this final sample batch can be seen in Figure 4 and ranged from 70–129%, with the 0.5 ng/mL (0.2 µg/g) spike of maize starch being the only sample to not sit within an acceptable recovery range. The total average recovery across all samples and spikes was 94%.
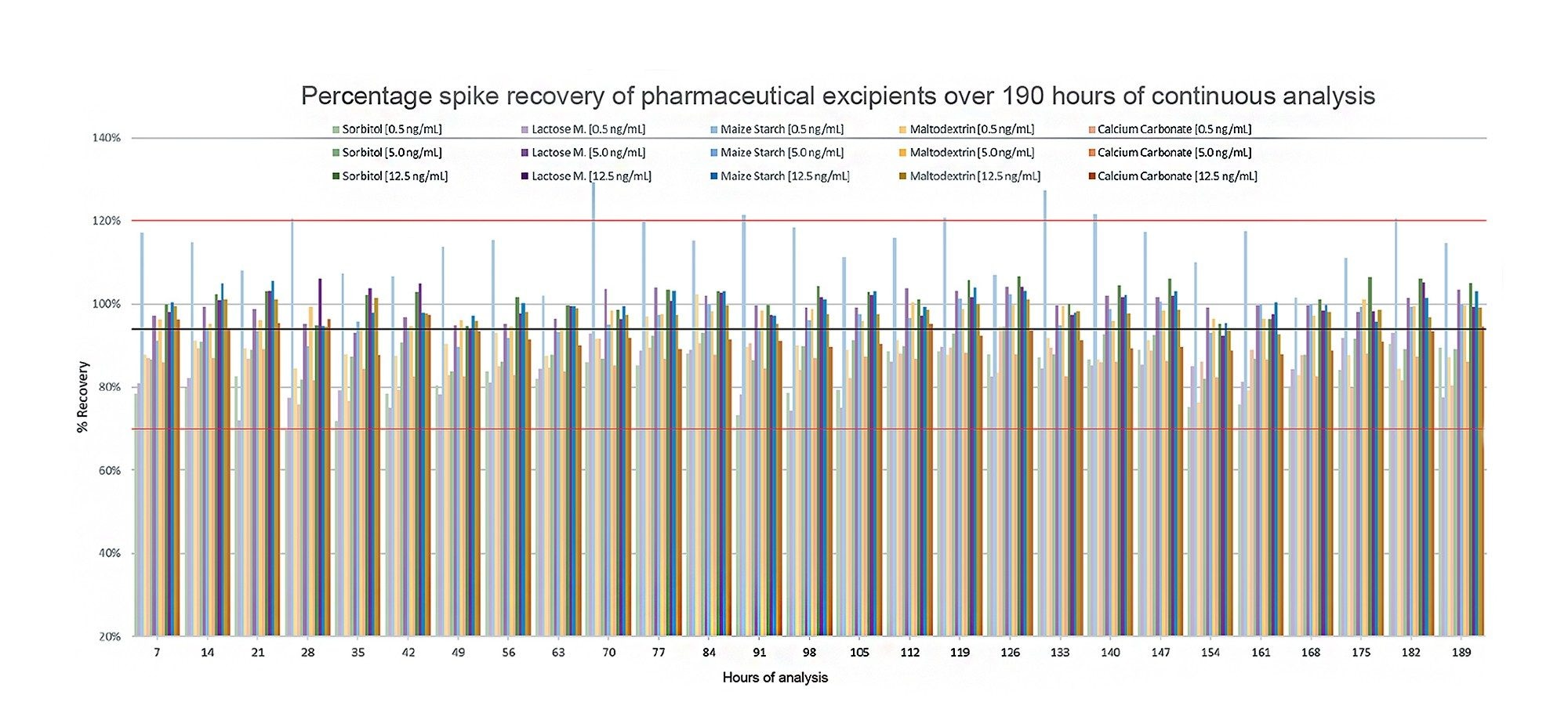 Figure 4. Nitrite recovery (%) in all spiked excipient samples across 190 hours of continuous acquisition. The red lines indicate a 70–120% tolerance interval, the black line indicates total average recovery of 94%.
Figure 4. Nitrite recovery (%) in all spiked excipient samples across 190 hours of continuous acquisition. The red lines indicate a 70–120% tolerance interval, the black line indicates total average recovery of 94%.
Figure 5 shows a representative example of absolute peak area for each of the unspiked excipients in a given sequence. Importantly, the process blank and excipient samples are prepared using the same sample preparation technique, therefore it is assumed that the intrinsic level of contamination present in the process blank is also present across all excipient samples. The level of NAT seen in the process blank in Figure 5 corresponds 0.041 ng/mL. As a result, this figure shows the relative absence of nitrite across all excipients analysed, with the exception of maize starch.
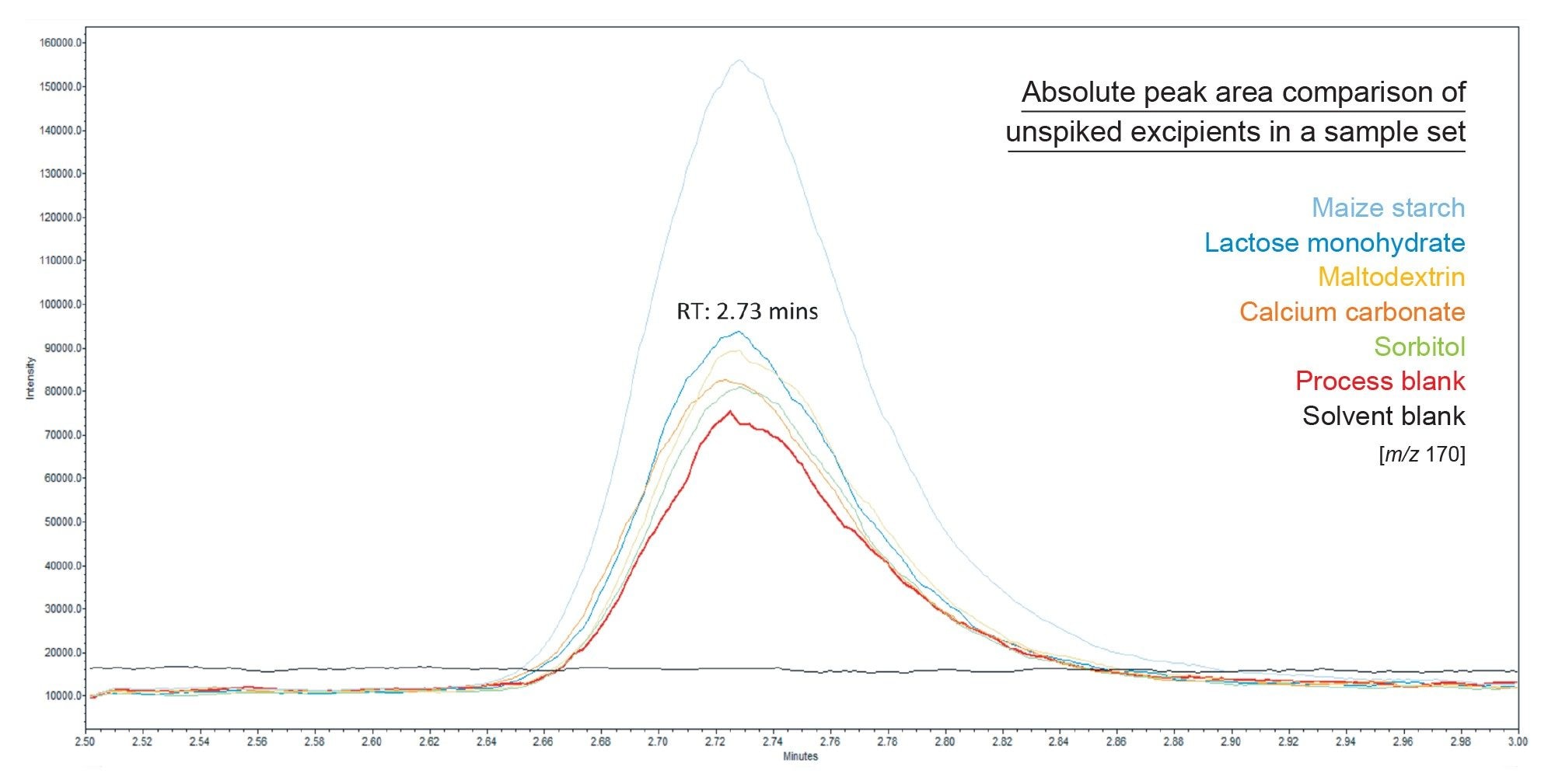 Figure 5. Overlayed SIR chromatograms of absolute peak area of nitrite in a process blank compared to unspiked excipient samples in a given sample sequence.
Figure 5. Overlayed SIR chromatograms of absolute peak area of nitrite in a process blank compared to unspiked excipient samples in a given sample sequence.
The presence of nitrite in the process blank, and not the MS sensitivity, is the limiting factor to this analysis. This presence arises from contamination that can originate from a variety of sources during sample preparation. The method is inherently reproducible, with %RSDs of the calculated value for the process blank ranging from 2.7 to 8.5% across the four sample batches acquired, however future efforts should focus on eliminating this contamination in order to lower the method limits of detection (LOD).
For the five excipients analysed over the course of the analysis, only maize starch was seen to contain levels of nitrite that would likely be flagged for further analysis. Figure 6 displays the reported concentration of an unspiked maize starch sample across 190 hours of analysis, which returned an average value for nitrite content of 0.14 µg/g, with a %RSD of 8.7% over n=27 injections. This value aligns with a consensus of mean values given for maize starch across excipient databases.6 The dotted line shown is an assumed example of a nominal reporting threshold for maize starch, which is plotted as 0.5x the reported mean value of nitrite content as described in literature. In this example, the excipient would exceed this threshold and be flagged for further analysis.
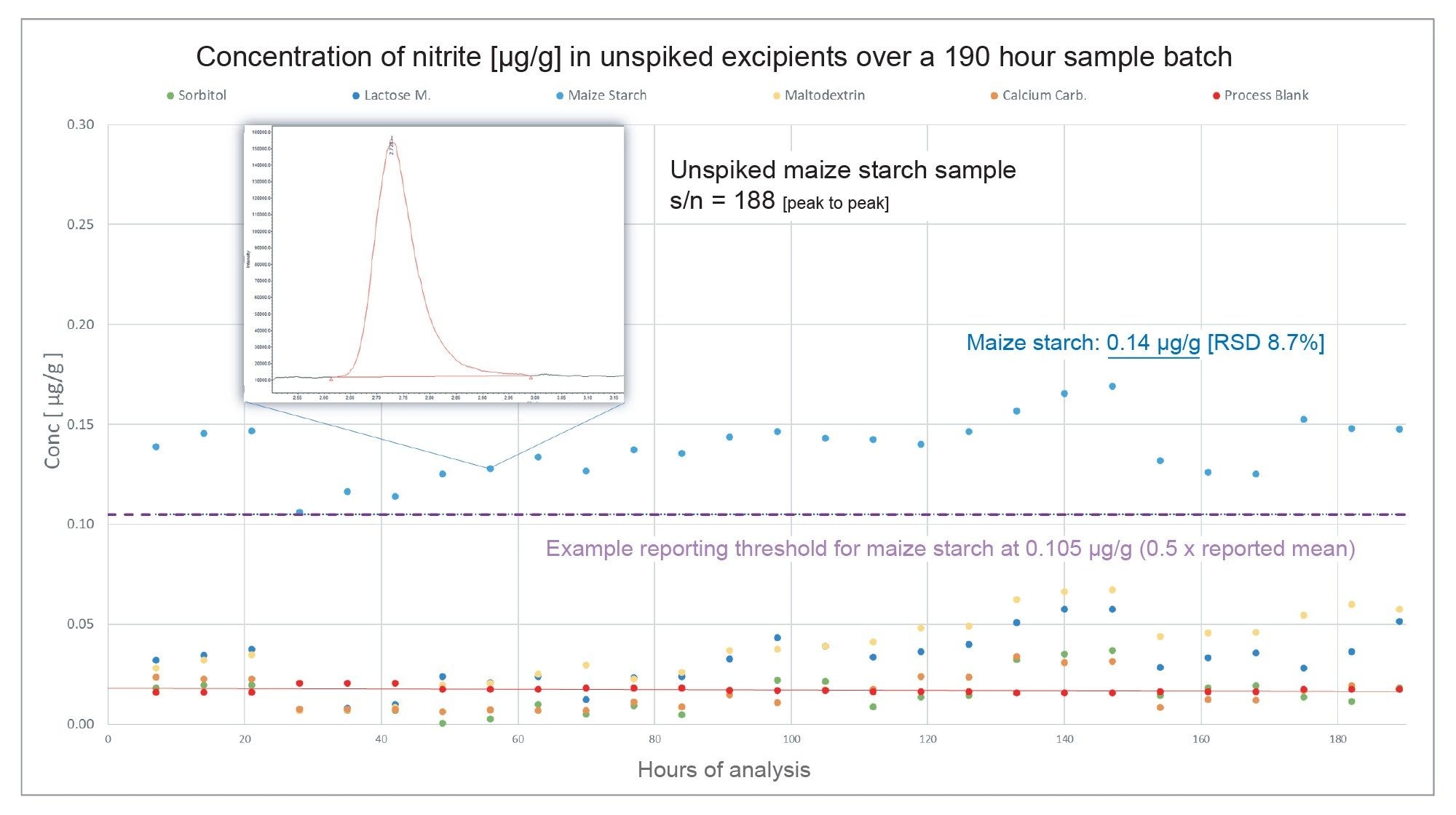 Figure 6. Nitrite content (µg/g) in unspiked excipient samples analysed over 190 hours of continuous acquisition. Nitrite content in the maize starch sample exceeded the literature reported 0.5 x mean value of 0.105 µg/g. The precision of measurement of nitrite in this sample over n=27 injections indicate an %RSD of 8.7%.
Figure 6. Nitrite content (µg/g) in unspiked excipient samples analysed over 190 hours of continuous acquisition. Nitrite content in the maize starch sample exceeded the literature reported 0.5 x mean value of 0.105 µg/g. The precision of measurement of nitrite in this sample over n=27 injections indicate an %RSD of 8.7%.
Contrastingly in Figure 7, using the same process to identify an example reporting threshold for lactose monohydrate, the excipient analysed did not exceed the 0.27 µg/g threshold and could therefore, in this instance, be considered acceptable for use.
The high variation in %RSDs here reflects the proximity of nitrite content in the excipient to that of the process blank. The relative difference in concentration between the two samples is very low, suggesting that there is no detectable nitrite content in the excipient. The high %RSD values are therefore a reflection of the limitations of the method, and not instrument performance.
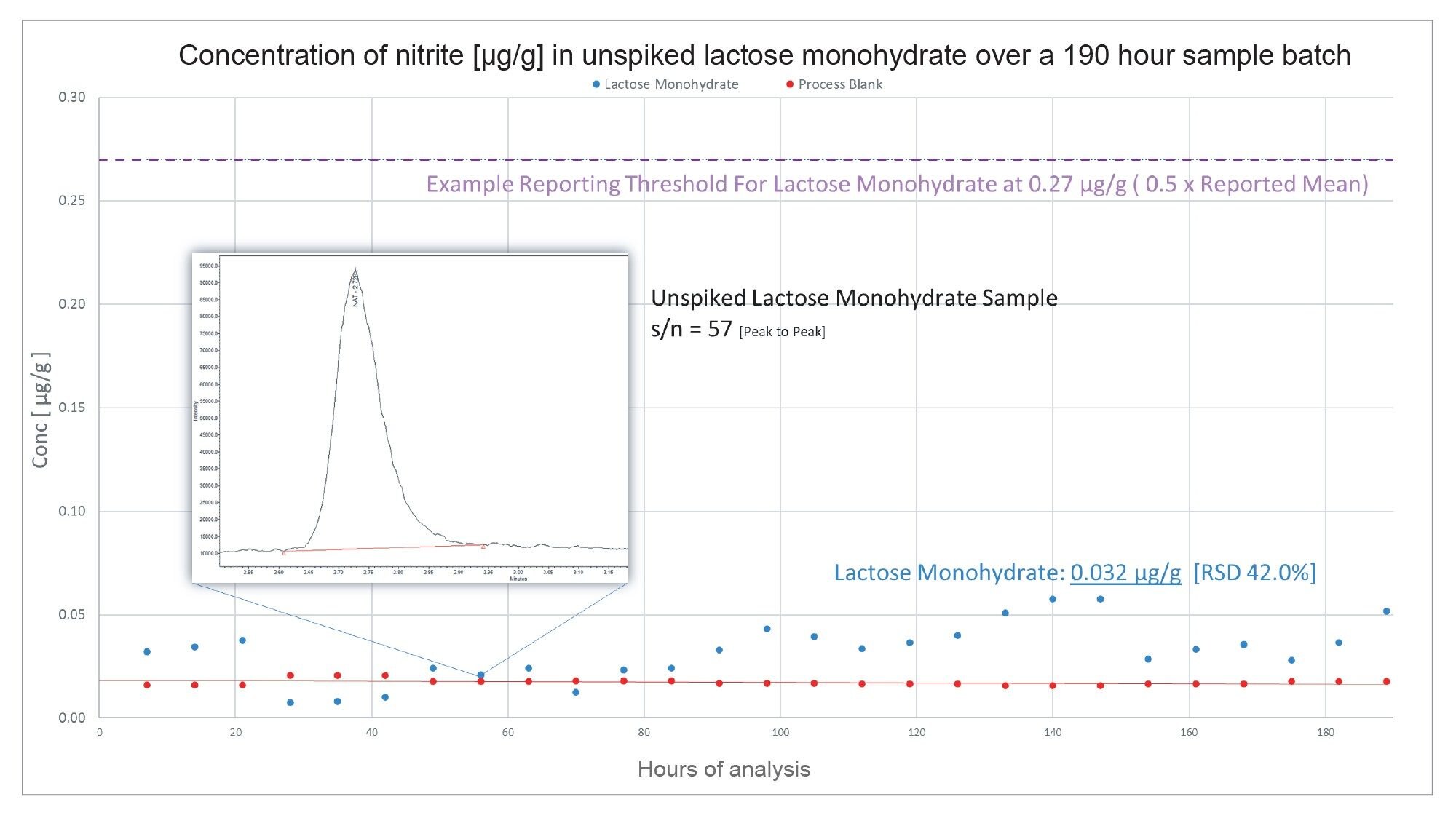 Figure 7. Nitrite content (µg/g) of unspiked lactose monohydrate over 190 hours of continuous analysis. Nitrite content in the lactose monohydrate sample did not exceed the literature reported 0.5 x mean value of 0.27 µg/g.
Figure 7. Nitrite content (µg/g) of unspiked lactose monohydrate over 190 hours of continuous analysis. Nitrite content in the lactose monohydrate sample did not exceed the literature reported 0.5 x mean value of 0.27 µg/g.
Other unspiked excipient values for a given sample batch can also be seen plotted in Figure 6. These values are also not significantly above the threshold of the process blank to be accurately quantified. Likewise, this infers that there is no presence of nitrite within these excipients, or that the level of nitrite is as low as the method limit of detection, and therefore these samples would not be flagged for further analysis.
In Figure 7, the signal to noise ratio of the unspiked lactose monohydrate sample suggests that the sensitivity of the method is limited by the process blank and not the capabilities of the instrument. The system sensitivity (as shown in the example chromatograms embedded in the above figures) is more than sufficient to detect levels just above (Figure 6) and well below (Figure 7) the reporting thresholds. Previous reported data suggests an LOD for the method of 0.0013 ng/mL.8
Using Empower CDS reporting functionality allows for simple reviewing of excipient nitrite concentration within a sample batch, as seen in Figure 8. This allows for fast and comprehensive decision making, while ensuring compliance and data integrity within your laboratory. In the example shown, an unspiked maize starch sample displays a reported concentration of 0.392 ng/mL, or 0.157 µg/g.
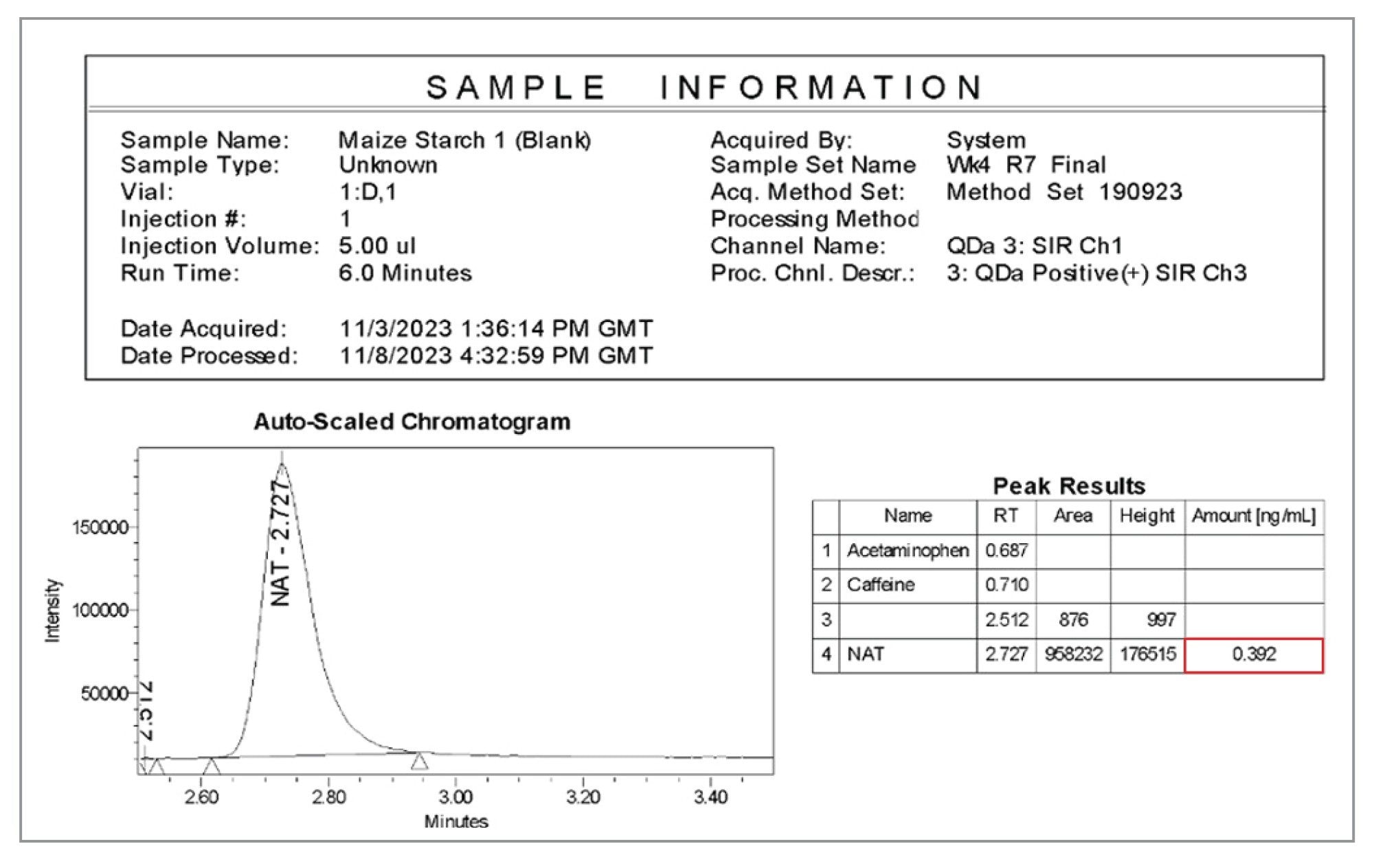 Figure 8. Example of Empower CDS generated data report for nitrite content in a maize starch sample.
Figure 8. Example of Empower CDS generated data report for nitrite content in a maize starch sample.
Instrument Robustness
Aside from method performance, overall instrument uptime and reliability were assessed, with instrument interaction and any maintenance procedures documented across the analysis. Over the course of 29 days, a total of 5792 injections were performed with the instrument, with 1920 of those being excipient samples. An overview of the analysis can be seen in Figure 9.
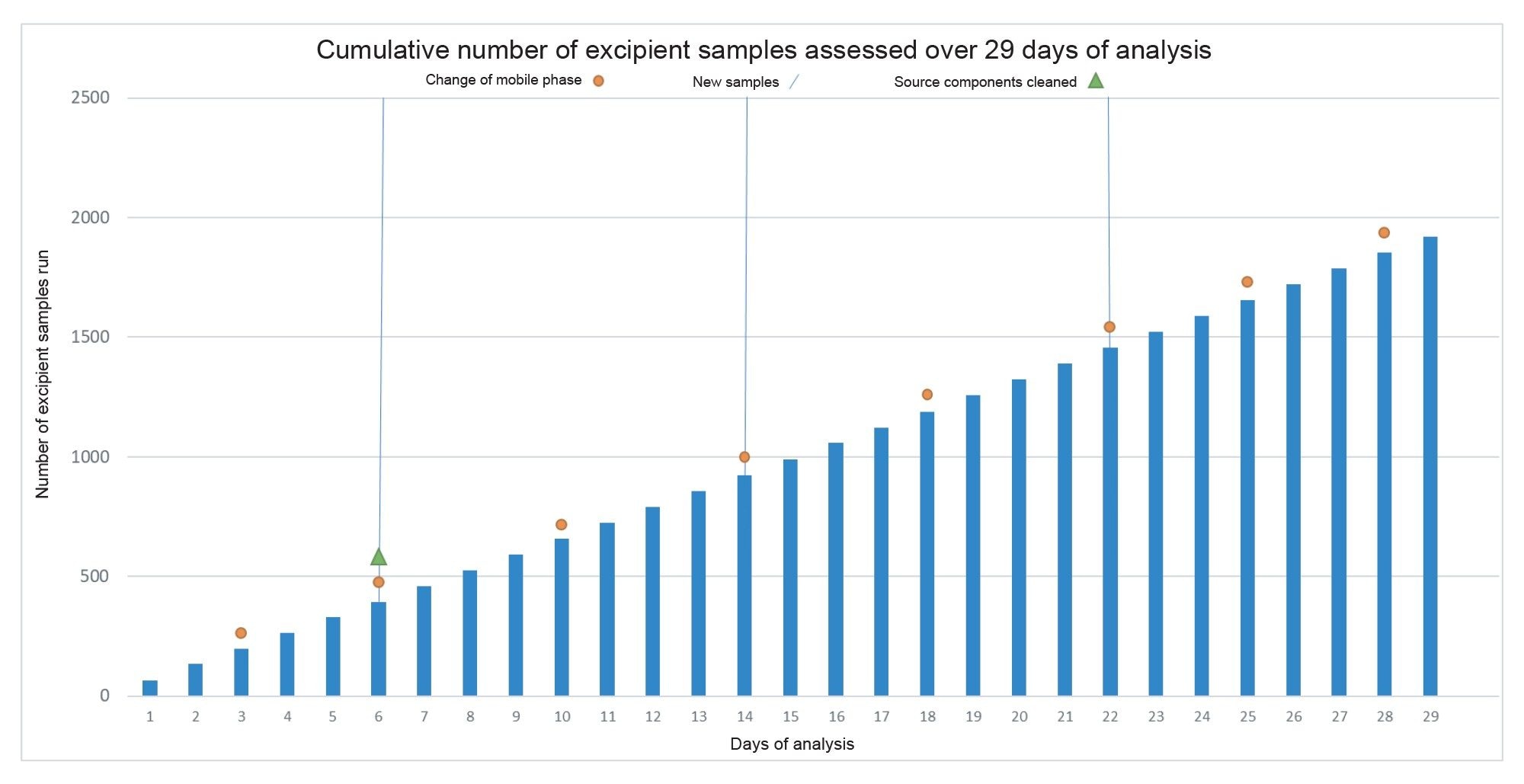 Figure 9. Cumulative plot of excipient samples analysed over 29 days of analysis - annotated to show mobile phase changes, introduction of new samples, and any cleaning required.
Figure 9. Cumulative plot of excipient samples analysed over 29 days of analysis - annotated to show mobile phase changes, introduction of new samples, and any cleaning required.
Limited system interaction was needed over the 29-day analysis period. During 676 hours of batch analysis, the instrument required only two hours of maintenance, involving a 20-minute venting and cleaning process. The system resumed analysis within two hours. Despite the source cleaning occurring after six days, the system remained fully operational for the subsequent 23 days, suggesting contamination was likely associated with prior instrument use. Additional forms of instrument interaction are detailed in Table 1.
Source components were cleaned once due to QC injections deviating from an acceptable range. Instrument resolution, tuning, and calibration were automated during start-up, requiring no analyst intervention.
Mobile phase and wash solutions were replenished as needed. No nitrite carryover was observed in any of the solvent blanks (n=1920 across the entire analysis), which raises the prospect of higher sample throughput using this method.
Robustness of the QDa II Mass Detector was a crucial metric to assess the viability of this workflow. Instrument QCs (n=768 per level) were observed over the course of the analysis to assess the instrument robustness independently from the nitrite samples. Results were consistent across the analysis. Within any given sample batch, %RSDs for a given QC level did not exceed 9.8%. In the 520 hours of continuous operation following the source component clean, %RSDs were <12.5% across both QC compounds and spike levels, as shown in Figure 10.
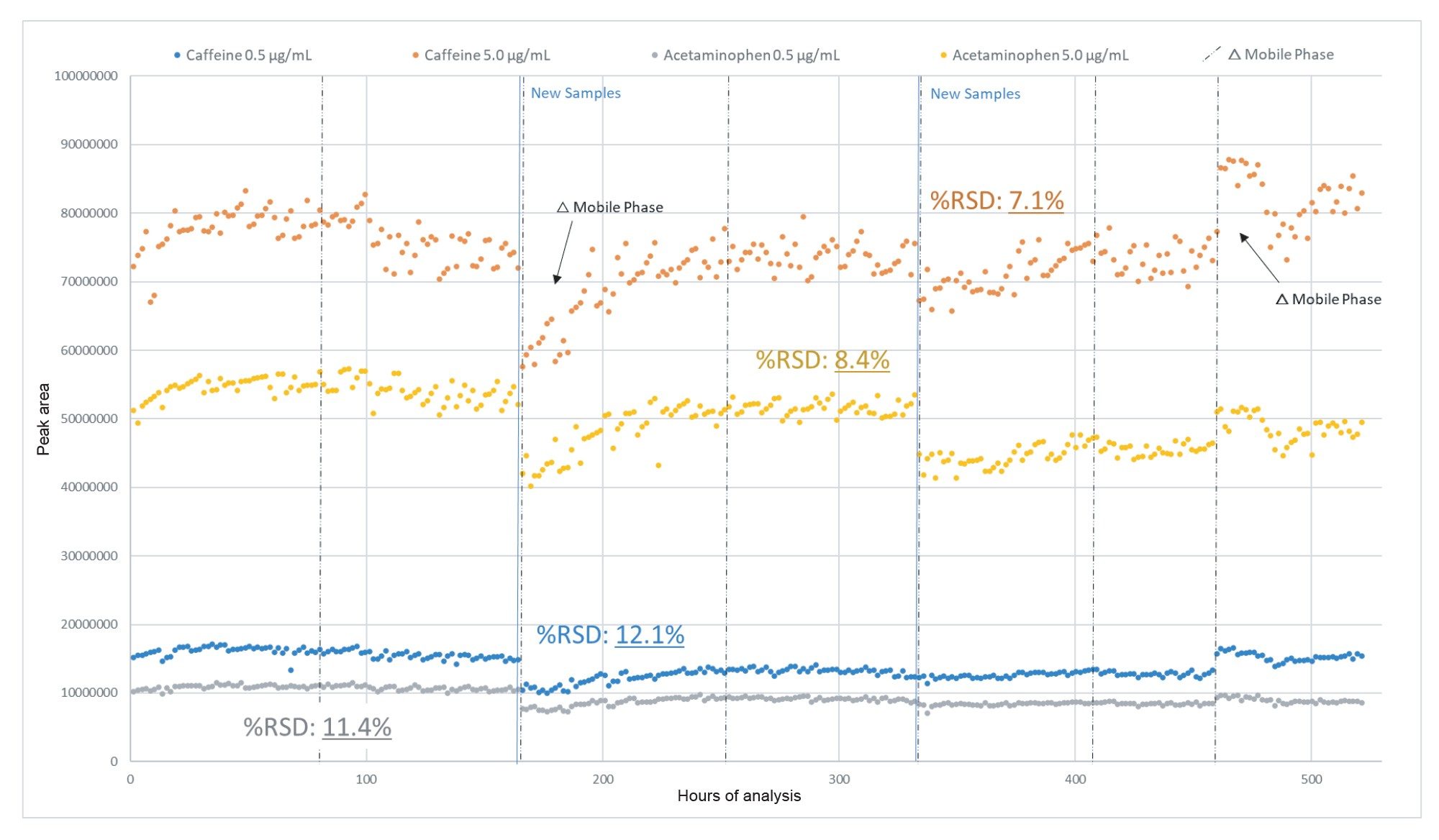 Figure 10. Absolute peak area of QC components (caffeine & acetaminophen) over 520 hours of continuous analysis (data points prior to sample cone clean excluded).
Figure 10. Absolute peak area of QC components (caffeine & acetaminophen) over 520 hours of continuous analysis (data points prior to sample cone clean excluded).
Instrument robustness can also be demonstrated across each sample batch by assessing %RSDs for absolute peak area of nitrite standard injections. Here, %RSDs did not exceed 14.7% within any given sample batch across the entirety of the analysis, as shown in Table 2.
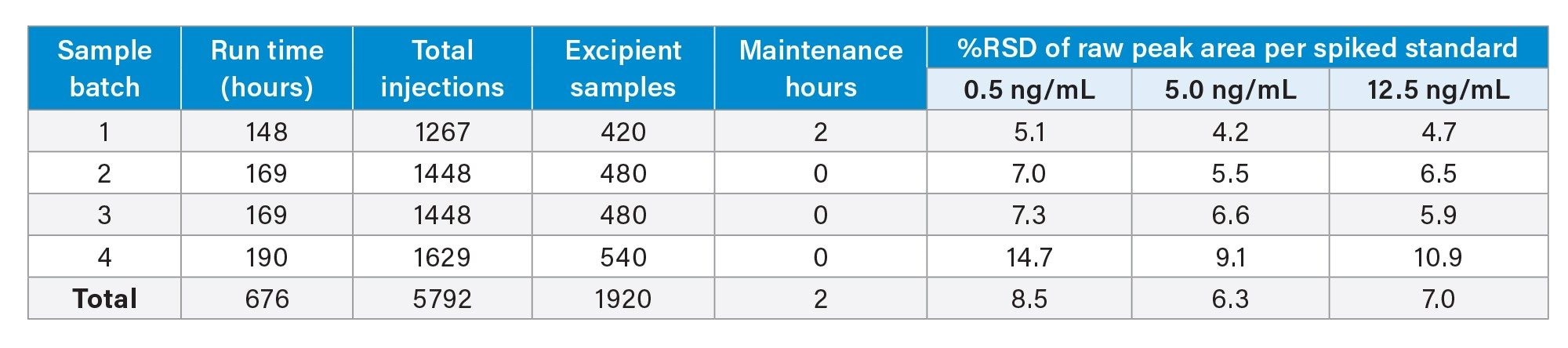 Table 2. A breakdown of the number of injections and excipient samples analysed over the course of the robustness study, as well as %RSDs of absolute peak area for spiked standards (n≥42 for any given spike level).
Table 2. A breakdown of the number of injections and excipient samples analysed over the course of the robustness study, as well as %RSDs of absolute peak area for spiked standards (n≥42 for any given spike level).
Reported concentrations across excipient spikes were mostly in line with expectations. Spiked excipients showed %RSDs that were <20% across all spike levels and excipient types throughout the analysis. An example can be seen in Figure 11, where %RSDs are shown for all spike levels of calcium carbonate throughout the analysis, not exceeding 13.9%.
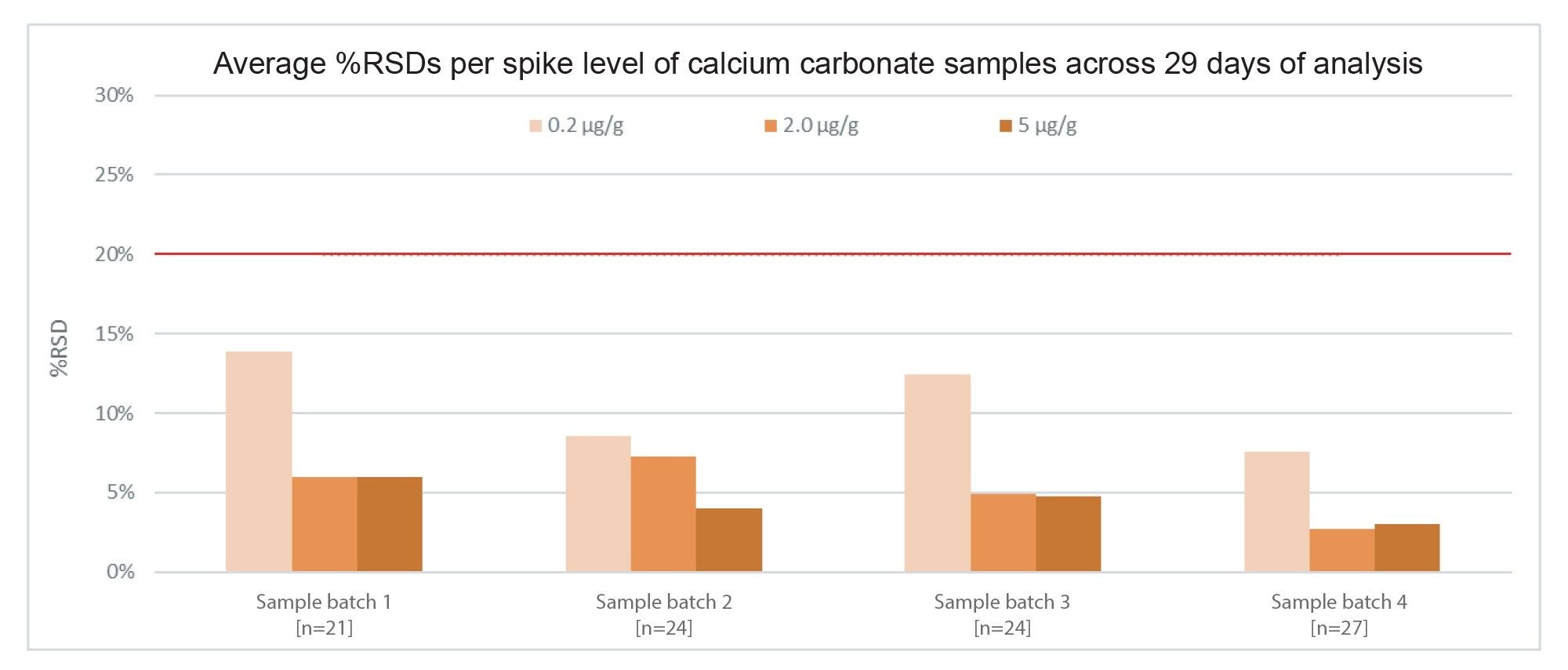 Figure 11. Average %RSDs of 0.2, 2.0, and 5.0 µg/g nitrite spikes in calcium carbonate excipient samples, across (n) replicates per spike level in a given sample batch. The red line indicates a %RSD threshold of 20%.
Figure 11. Average %RSDs of 0.2, 2.0, and 5.0 µg/g nitrite spikes in calcium carbonate excipient samples, across (n) replicates per spike level in a given sample batch. The red line indicates a %RSD threshold of 20%.
Shown in Figure 12 are the average %RSD values for each excipient sample and spike level over the course of the 29 days of analysis. The %RSDs across the 0.2, 2.0, and 5.0 µg/g (ppm) spike levels of the five excipients analysed across the entirety of the analysis did not exceed 18.5%, 11.1%, and 6.0% respectively, demonstrating robust instrument performance over time.
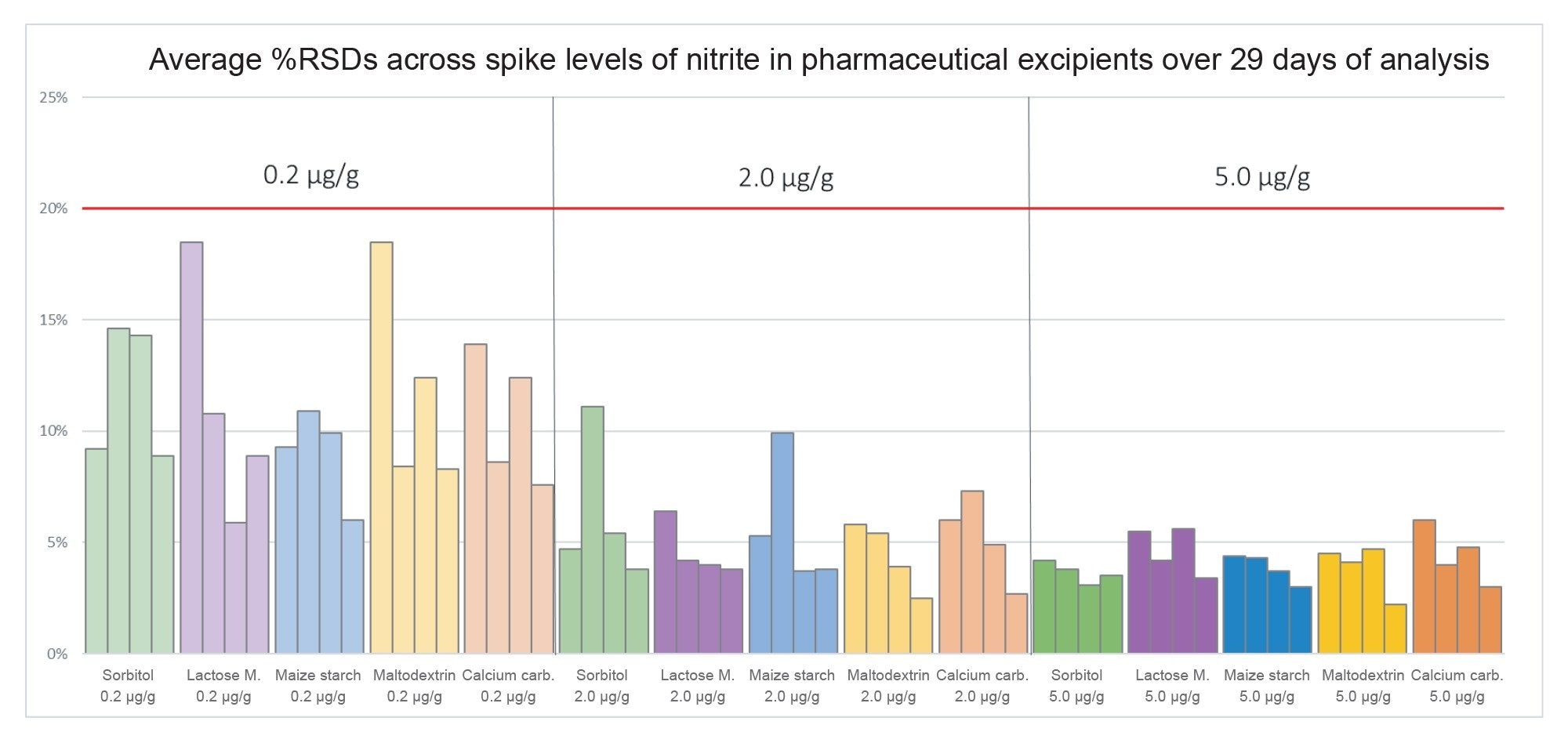 Figure 12. Average %RSDs of all excipient spike levels throughout the analysis, where each of the four datapoints per excipient spike level represents an average value for a spike level in a given sample batch (N.B. one replicate was excluded from 0.2 ug/g maltodextrin spike level in Sample Batch 1, due to error in sample preparation).
Figure 12. Average %RSDs of all excipient spike levels throughout the analysis, where each of the four datapoints per excipient spike level represents an average value for a spike level in a given sample batch (N.B. one replicate was excluded from 0.2 ug/g maltodextrin spike level in Sample Batch 1, due to error in sample preparation).
Conclusion
- The results from the experiments performed indicate that the ACQUITY QDa II Mass Detector is a robust and reliable instrument suitable for routine high throughput applications, such as the analysis of nitrite in pharmaceutical excipients
- The instrument successfully analysed ~6000 injections over 676 hours of analysis, experiencing only 2 hours of downtime
- The %RSD of instrument QC injections in a given sample batch did not exceed 9.8%, demonstrating consistent performance
- Throughout 520 hours of continuous analysis, the %RSD of absolute peak area for any given QC level remained below 12.5%, demonstrating excellent stability of the system throughout the analysis
- The %RSDs of absolute peak area for all nitrite standard injections was consistently less than 14.7% for any given sample batch analysed
- Nitrite levels were successfully detected in maize starch excipients at 0.14 µg/g, in line with mean levels reported in excipients databases, indicating the quantitative accuracy of the method. Other excipients did not exhibit detectable levels of nitrite during the analysis
- This 6-minute method using the ACQUITY Arc UHPLC and ACQUITY QDa II Mass Detector is an efficient and cost-effective approach for µg/g level quantitation of nitrite in pharmaceutical excipient analysis
References
- Brambilla, G., & Martelli, A. (2007). Genotoxic and carcinogenic risk to humans of drug-nitrite interaction products. In Mutation Research - Reviews in Mutation Research (Vol. 635, Issue 1, pp. 17–52). https://doi.org/10.1016/j.mrrev.2006.09.003.
- Horne, S., Vera, M. D., Nagavelli, L. R., Sayeed, V. A., Heckman, L., Johnson, D., Berger, D., Yip, Y. Y., Krahn, C. L., Sizukusa, L. O., Rocha, N. F. M., Bream, R. N., Ludwig, J., Keire, D. A., & Condran, G. (2023). Regulatory Experiences with Root Causes and Risk Factors for Nitrosamine Impurities in Pharmaceuticals. In Journal of Pharmaceutical Sciences (Vol. 112, Issue 5, pp. 1166–1182). Elsevier B.V. https://doi.org/10.1016/j.xphs.2022.12.022.
- Cioc, R. C., Joyce, C., Mayr, M., & Bream, R. N. (2023). Formation of N-Nitrosamine Drug Substance Related Impurities in Medicines: A Regulatory Perspective on Risk Factors and Mitigation Strategies. In Organic Process Research and Development. American Chemical Society. https://doi.org/10.1021/acs.oprd.3c00153.
- Wu, Y., Levons, J., Narang, A. S., Raghavan, K., & Rao, V. M. (2011). Reactive impurities in excipients: Profiling, identification and mitigation of drug-excipient incompatibility. In AAPS PharmSciTech (Vol. 12, Issue 4, pp. 1248–1263). https://doi.org/10.1208/s12249-011-9677-z.
- Berardi, A., Jaspers, M., & Dickhoff, B. H. J. (2023). Modeling the Impact of Excipients Selection on Nitrosamine Formation towards Risk Mitigation. Pharmaceutics, 15(8). https://doi.org/10.3390/pharmaceutics15082015.
- Boetzel, R., Schlingemann, J., Hickert, S., Korn, C., Kocks, G., Luck, B., Blom, G., Harrison, M., François, M., Allain, L., Wu, Y., & Bousraf, Y. (2023). A Nitrite Excipient Database: A Useful Tool to Support N-Nitrosamine Risk Assessments for Drug Products. Journal of Pharmaceutical Sciences, 112(6), 1615–1624. https://doi.org/10.1016/j.xphs.2022.04.016.
- Wang, Q. H., Yu, L. J., Liu, Y., Lin, L., Lu, R. gang, Zhu, J. ping, He, L., & Lu, Z. L. (2017). Methods for the detection and determination of nitrite and nitrate: A review. In Talanta (Vol. 165, pp. 709–720). https://doi.org/10.1016/j.talanta.2016.12.044.
- Jireš, J., & Douša, M. (2022). Nitrites as precursors of N-nitrosation in pharmaceutical samples – A trace level analysis. Journal of Pharmaceutical and Biomedical Analysis, 213. https://doi.org/10.1016/j.jpba.2022.114677.
Featured Products
720008197, February 2024