CRISPR Single Guide RNA Characterization by IP- RP-LC-MS with a Premier Oligonucleotide BEH 300 Å C18 Column
Abstract
The high speed, low cost, and reduced need for animal use has made CRISPR/Cas systems the go-to platform for genome editing in both research and therapeutics. With this system, a genome can be altered at precise regions of genomic DNA, programmable by an independent single guide RNA sequence. Once bound, a CRISPR ribonucleoprotein complex will subsequently cleave, alter and/or modify the target locus. The specificity of the editing is directly tied to the design and quality of the loaded RNA molecule.
Advanced analytical methods involving ion-pairing reversed phase liquid chromatography coupled with mass spectrometry (IP-RP-LC-MS) can help ensure the identity, purity, integrity, and intended modifications of the CRISPR RNA. A column constructed with high-performance surfaces and oligonucleotide batch tested WidePore BEH C18 sorbent is proposed as a starting point for a reliable separation of both intact and digested sgRNA. The resolution offered by this column makes it possible to perform direct molecular analysis through intact mass measurements and MSE-based sequencing of digested components.
Benefits
- Premier Oligonucleotide BEH C18 300 Å Columns are well suited to the analysis of both intact sgRNA and its nuclease digested sgRNA components
- Chromatographic resolution to distinguish failure sequences and degradants of intact RNA for robust integrity and purity assessment
- Excellent IP-RP-LC separation of digestion components and modified termini for detailed molecular characterization
- Accurate mass matching and sgRNA oligo mapping annotation through the use of in silico digestion calculator, waters_connect™, UNIFI™, and CONFIRM Sequence applications
Introduction
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) is a genome editing system that was first discovered in bacteria as an adaptive immune response against invading bacteriophages.1 A naturally occurring CRISPR sequence consists of tracrRNA (trans-activating CRISPR RNA), and a short piece of RNA, called crRNA.2 These two oligonucleotides concatenate into a single guide RNA (sgRNA). Besides RNA, the complex contains a Cas protein, such as Cas9, which is an endonuclease that works concert with the sgRNA to introduce a double stranded break in target DNA for programmable genome editing (Figure 1).3 Editing by a CRISPR/Cas-9 ribonucleoprotein (RNP) system occurs in three important steps – (i) genome target site recognition, (ii) cleavage of both strands, and (iii) repair.4 Accuracy and effectiveness of editing depends on the ability of sgRNA to uniquely target the desired location of the gene locus that is to be altered. Incorrect matching can lead to off-target effects and loss of efficiency.5 Significant amounts of interest in CRISPR research led to the engineering of multiple Cas proteins as well as several clinical trials aimed at treating diseases with ex vivo gene edited cell therapies and in vivo gene editing.6,7
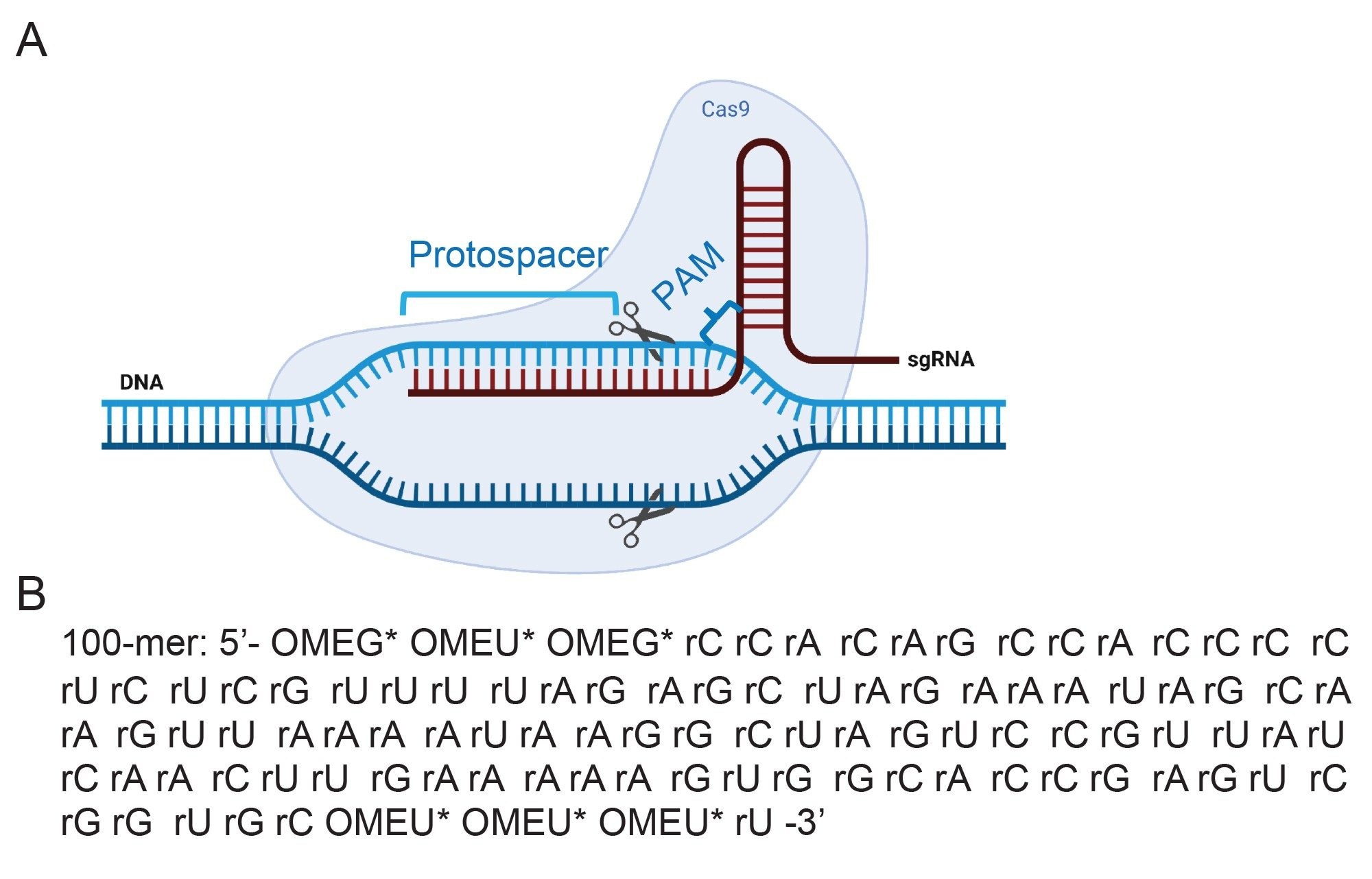 Figure 1. (A) A schematic representation of a CRISPR-Cas9 ribonucleoprotein (RNP) complex with a single guide RNA (sgRNA) bound to a Cas9 protein and complementary DNA. Created with BioRender.com.
Figure 1. (A) A schematic representation of a CRISPR-Cas9 ribonucleoprotein (RNP) complex with a single guide RNA (sgRNA) bound to a Cas9 protein and complementary DNA. Created with BioRender.com.(B) Sequence of the synthetic sgRNA of interest in this analysis. OME= 2’-methyl, * denotes phosphorothioate linker. The sgRNA directs Cas-9 to recognize the target DNA sequence (protospacer) through 5’ crRNA and cuts the DNA at a region upstream of PAM sequence (5’-NGG-3’, N can be any nucleotide base) after DNA melting and RNA-DNA hybrid formation.
Because of the potential for off-target effects, the sequence, modifications, and impurities of an sgRNA should be very carefully characterized and monitored. To this end, intact RNA analysis, oligo mapping and sequencing can be performed through liquid chromatography and mass spectrometry (LC-MS). These advanced methods provide direct molecular observations of the RNA sequence as opposed to the indirect information obtained by complementary DNA (cDNA)-based Sanger and next-generation sequencing (NGS), where modification information is lost. We report the utilization of an ACQUITY Premier BEH C18 300 Å, 1.7 µm Column for ion-pairing reversed phase LC of both intact and digested RNA using a single platform comprised of LC, UV, and MS instruments with application-based software for data acquisition, processing, and reporting of results. The high-performance surface technology and WidePore BEH particles contained in this column ensure the quick start up of new separations and reliable resolution of intact sgRNA. When used with an oligo mapping approach, this column has also helped to identify phosphorothioate variants. In this work, IP-RP-LC-MS data were quickly annotated through the use of an in silico digestion library calculator (Waters Microapp Store) in combination with waters_connect™, its integrated UNIFI, CONFIRM sequence, and INTACT Mass applications.
Experimental
Intact sgRNA analysis
Mouse GATA2 sgRNA (Integrated DNA Technologies, Coralville, Iowa; 5’- OMEG* OMEU* OMEG* rC rC rA rC rA rG rC rC rA rC rC rC rC rU rC rU rC rG rU rU rU rU rA rG rA rG rC rU rA rG rA rA rA rU rA rG rC rA rA rG rU rU rA rA rA rA rU rA rA rG rG rC rU rA rG rU rC rC rG rU rU rA rU rC rA rA rC rU rU rG rA rA rA rA rA rG rU rG rG rC rA rC rC rG rA rG rU rC rG rG rU rG rC OMEU* OMEU* OMEU* rU -3’, OME=2’-O-methyl, *=phosphorothioate) was resuspended in nuclease-free Tris-EDTA buffer to prepare a 10 pmol/μL sample solution.
LC Conditions
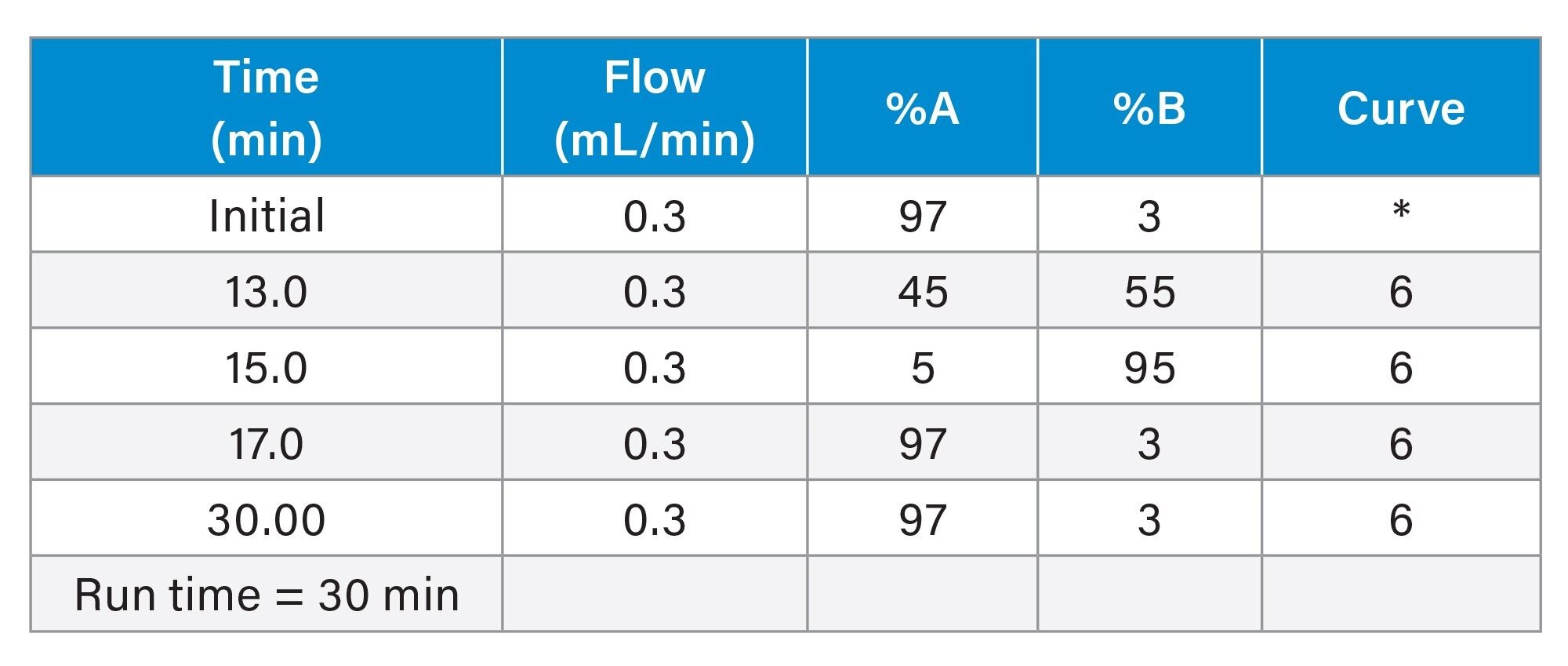
|
LC system: |
ACQUITY UPLC I-ClassFTN (as part of the BioAccord™ System) |
|
Detector: |
ACQUITY UPLC TUV Detector |
|
Wavelength: |
260 and 280 nm |
|
Column: |
ACQUITY Premier Oligonucleotide BEH C18, 300 Å, 1.7 µm, 2.1 x 100 mm (p/n: 186010540) |
|
Column temperature.: |
70 ˚C |
|
Sample temperature.: |
4 ˚C |
|
Injection: |
1 μL (10 pmol) |
|
Flow rate: |
0.3 mL/min |
|
Mobile phase A: |
0.1% N,N-diisopropylethylamine (DIPEA) as the IP reagent and 1% 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) in deionized water |
|
Mobile phase B: |
0.0375% DIPEA and 0.075% HFIP in 65:35 acetonitrile/water |
Gradient Table
MS Conditions
|
MS system: |
BioAccord LC-MS System |
|
Detector: |
ACQUITY RDa Detector |
|
Mode: |
Full scan with fragmentation |
|
Polarity: |
Negative |
|
Cone voltage: |
40 V |
|
Fragmentation cone voltage: |
80–200 V |
|
Mass range: |
High (400–5000 m/z) |
|
Scan rate: |
2 Hz |
|
Capillary voltage: |
0.80 kV |
|
Desolvation temperature: |
400 °C |
Sample preparation for Oligo mapping
Approximately 90 µg of mouse GATA2 sgRNA (IDT) was digested using 24 µg (~10 kU) of guanosine-specific ribonuclease T1 (Worthington Biochemical Corporation, Lakewood, NJ). First, the RNA was denatured with 20 µL of 8 M urea prepared in nuclease-free buffer (10 mM Tris-HCl pH 7.5, 0.1 mM EDTA) at 80 °C for five minutes. After cooling to room temperature, RNase T1 resuspended in nuclease-free buffer was added to the denatured RNA and incubated at 37 °C for 30 minutes. The digest was brought to 80 µL after the addition of 40 µL of nuclease free buffer and transferred to a polypropylene autosampler vial (300 µL, p/n: 186002639) for IP-RP-LC-MS analysis using a 30 K resolution Q-Tof mass spectrometer (Waters Vion MS).
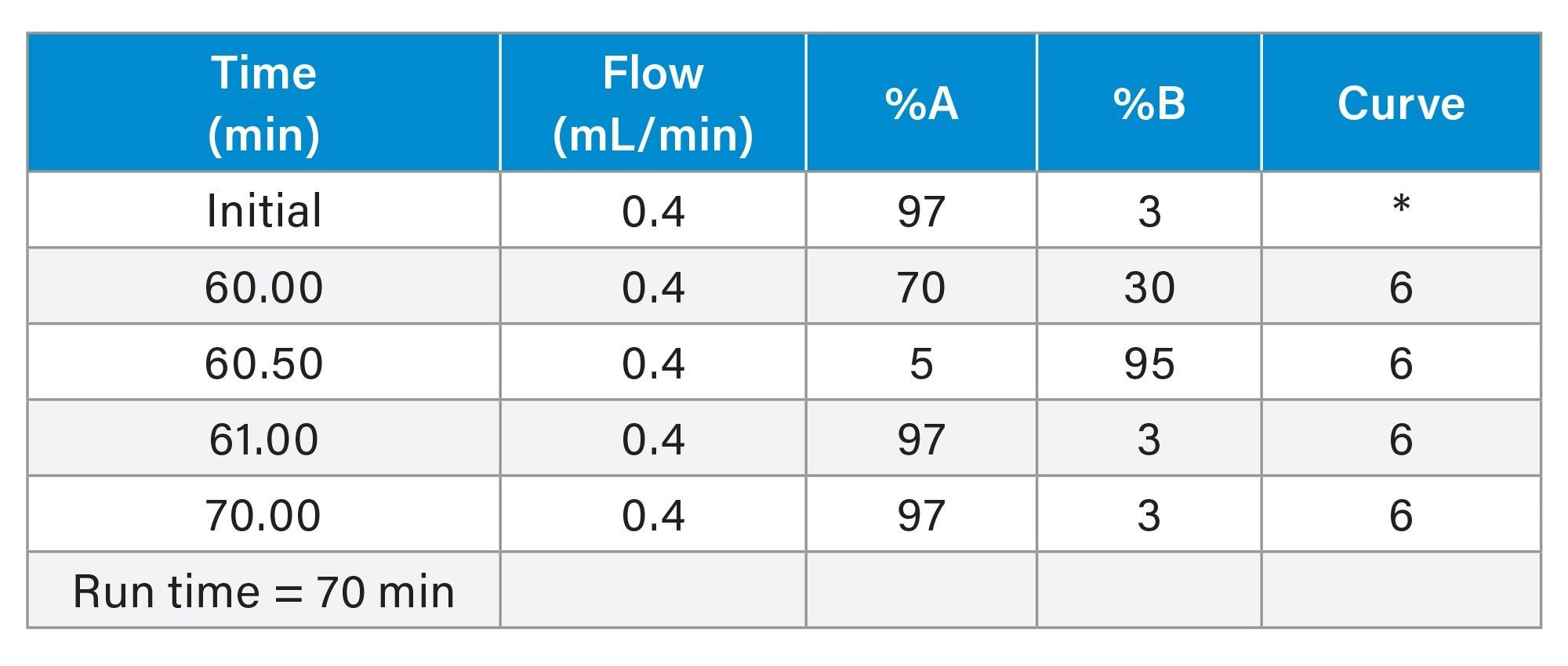
LC Conditions
|
LC system: |
ACQUITY UPLC Premier BSM System (as part of the BioAccord System) |
|
Detector: |
ACQUITY UPLC TUV Detector |
|
Wavelength: |
260 nm |
|
Column: |
ACQUITY Premier Oligonucleotide BEH C18, 300 Å, 1.7 µm, 2.1 x 150 mm (p/n: 186010541) |
|
Column temperature: |
70 ˚C |
|
Sample temperature: |
4 ˚C |
|
Injection: |
5 μL |
|
Flow Rate: |
0.4 mL/min |
|
Mobile phase A: |
0.1% N,N-diisopropylethylamine (DIPEA) as the ion-pairing (IP) reagent and 1% 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) in deionized water |
|
Mobile phase B: |
0.0375% DIPEA and 0.075% HFIP in 65:35 acetonitrile/water |
Gradient Table
MS Conditions
|
MS system: |
Vion MS system |
|
Mode: |
Full scan with fragmentation |
|
Polarity: |
Negative |
|
Cone voltage: |
80 V |
|
Capillary: |
1.5 kV |
|
Source temperature: |
150 °C |
|
Cone gas: |
50 L/h |
|
Desolvation gas: |
1200 L/h |
|
Desolvation temperature: |
600 °C |
|
Low collision energy: |
6 V |
|
Mass range: |
High (50–4000 m/z) |
|
Scan rate: |
2 Hz |
|
High collision energy ramp start: |
10 V |
|
High collision energy ramp end: |
30 V |
|
Fragmentation cone voltage: |
80–200 V |
|
Enable intelligent data capture: |
yes |
Informatics
The mRNA digestion calculator can be accessed via Waters Microapp Store, Intact Mass and Confirm Sequence applications are integrated with the waters_connect.
Results and Discussion
Intact sgRNA was injected onto an ACQUITY Premier Oligonucleotide BEH C18 Column (2.1 x 150 mm, 300 Å, 1.7 µm) and separated according to IP-RP-LC with a relatively short gradient using DIPEA/HFIP modified mobile phase. The mass spectrometer was programmed to acquire low energy mass spectra during this run for conducting intact RNA mass analysis. Data were also acquired in negative ion mode in triplicate with a BioAccord LC-MS System containing an RDa high resolution mass analyzer and detector. The applied column provides a WidePore stationary phase that is optimized for the analysis of nucleic acids up to 200 nt in length, which makes it highly suitable for sgRNA molecules that are typically around ~100 nucleotides long. As shown in Figure 2, the total ion chromatogram (TIC) profile (spanning from 2 to 5.5 minutes) using the 300 Å BEH C18 column resolved various other sample components in addition to the main RNA component. The mass spectrum of the base peak at 4.95 minutes revealed an envelope of multiply charged ions. Deconvolution by the waters_connect intact mass application revealed a molecular weight of 32,242 Da which is very close to the expected mass value of 32,240 Da provided by the vendor. This chromatography also resolved several components such as failure sequences or shorter versions of sgRNA (with shorter retention time) and potential adducts/extensions from the intact sgRNA. Thus, the separation appeared to be well suited to checking the purity, integrity, separation, and eventual identification of impurities present in the synthesized sgRNA sample.
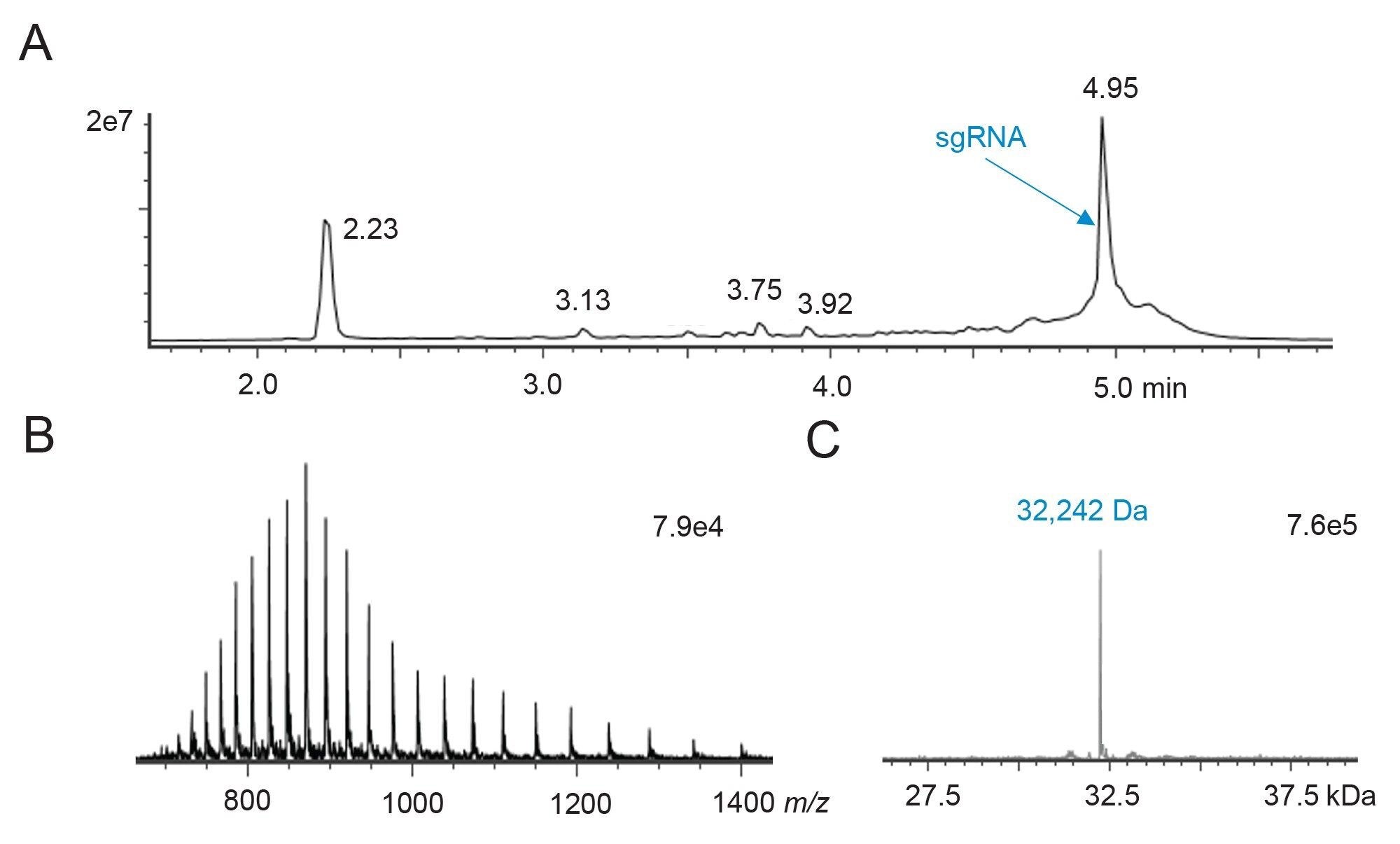 Figure 2. IP-RP-LC-MS analysis of a 10 pmol quantity of intact sgRNA.
Figure 2. IP-RP-LC-MS analysis of a 10 pmol quantity of intact sgRNA.(A) Total ion chromatogram as acquired with an ACQUITY PREMIER Oligonucleotide BEH C18 300 Å, 1.7 µm 2.1 x 100 mm column and a BioAccord system.
(B) Raw ESI negative ion mode mass spectrum for the peak at retention time 4.95 min.
(C) Deconvoluted mass spectrum and observed molecular weight, 32,242 Da (compared to the vendor reported mw of 32240 Da) for the intact sgRNA.
To date, it has been preferential to manufacture sgRNAs by synthetic means. Further, to improve resistance against degradation, it is common practice to include phosphorothioate (indicated by *) nucleotides with 2’-O-methyl groups (OME) at their 5’- and 3’-ends. We investigated the ability of IP-RP-LC-MS to identify and track these types of modifications. RNase T1 digestion and oligo mapping was applied in a way that has been previously described [8].8 The informatics approach started with in silico mRNA digestion calculator (Waters Microapp Store) and its use to predict the oligonucleotide digestion products corresponding to an RNase T1 digest. Several parameters including ‘no modification’, enzyme (RNase T1) and allowable missed cleavages (one or zero) were specified. The calculator provided default values for charge states based on m/z range, monoisotopic or average mass values. The csv file produced by the calculator was added to UNIFI, waters_connect software, and the CONFIRM Sequence application to facilitate different levels of peak identification. Phosphorothioate and 2’-O-methyl group containing 5’ and 3’-terminal oligonucleotides were predicted and manually added to the search space using the CONFIRM Sequence application. In sum, a library of individual analyte digestion products was assembled and imported into the UNIFI Scientific Library. Along with low energy ion scans, the MS was programmed to acquire high energy fragmentation ion spectra to corroborate LC-peak identifications. A 30 K mass resolution QTof instrument was used in this stage of work. Figure 3 shows a TIC profile for the sgRNA digested with RNase T1. The resulting oligonucleotide digestion products exhibited peaks with minimal retention time variation (≤0.01 min) over triplicate injections. All the expected digestion products eluted between three and 47 minutes. The HRMS Screening Analysis Method available within UNIFI was applied to identify oligonucleotide digestion products by screening the mass spectra and matching with the library of analytes within the defined tolerance limits. The mass spectral matches were manually verified to check on mass accuracy, charge states and the presence of sequence-informative fragment ions.
Oligonucleotide digestion components exclusively containing phosphodiester backbones exhibited sharp and symmetrical peaks, while the digestion components containing phosphorothioate bonds were observed as split peaks as shown in Figure 3B and 4. Extracted ion chromatograms for these 5’ and 3’ termini species confirmed that they were uniquely resolved into multipeak patterns, presumably due to diastereoisomer separations. To confirm the sequence of each oligomer, the LC-MS data was processed using the CONFIRM Sequence application where a minimum of 500 intensity counts was considered for scoring the presence of sequence-informative product ions in the MSE spectra. Figure 5 depicts the identification of the 5’-end and 3’-end digestion components of the sgRNA, including high confidence mass matching of fragment ions that could be used to localize and count the number of phosphorothioate groups. Dotmap representations of these matches further illustrates the observed fragment ions.
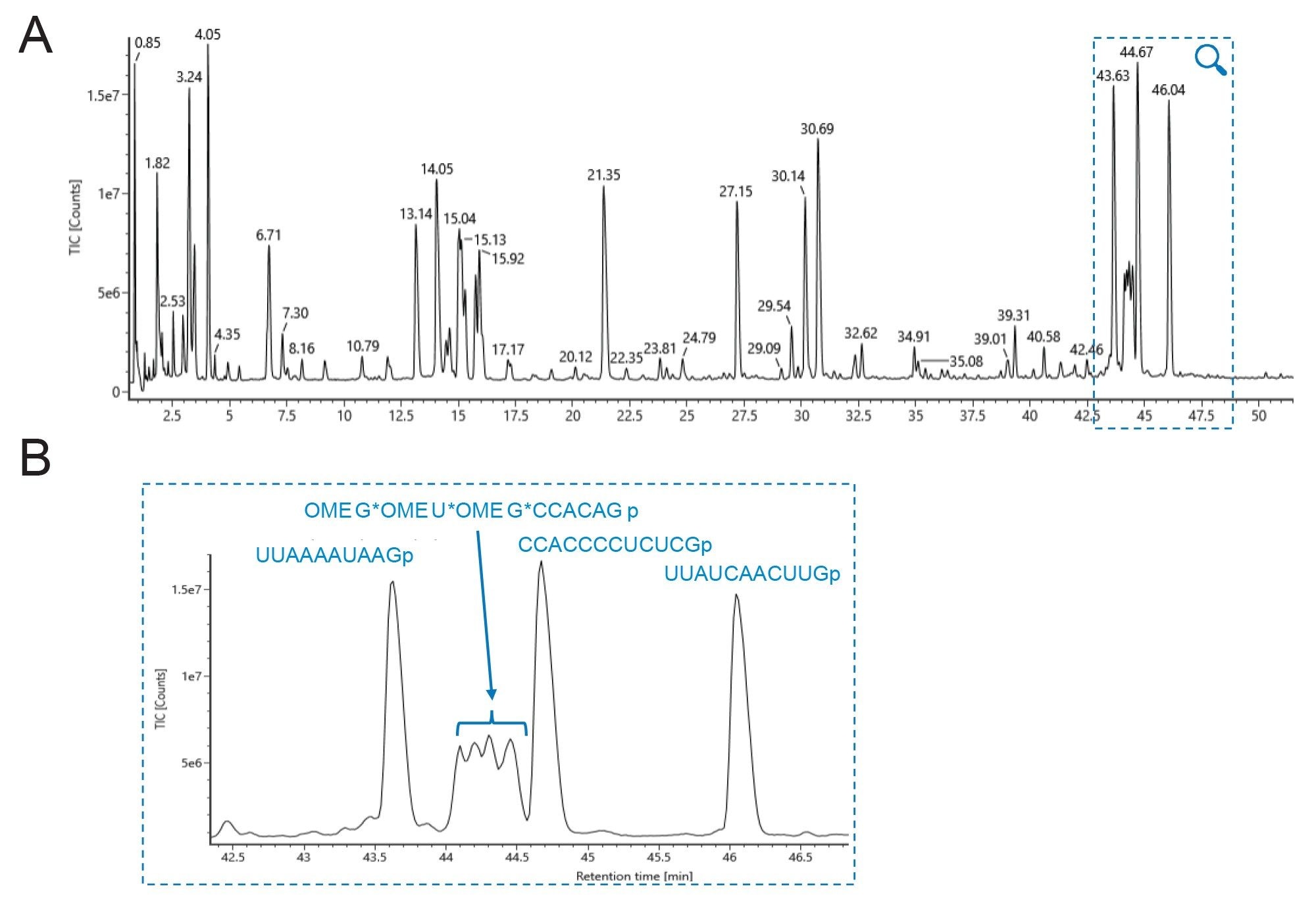 Figure 3. Oligo mapping of an RNase T1 digested sgRNA by IP-RP-LC-MS with an ACQUITY PREMIER Oligonucleotide BEH C18 300 Å, 1.7 µm 2.1 x 100 mm column as hyphenated with UV and QTof HRMS detection. (A) Full view of IP-RPLC total ion chromatogram (TIC).
Figure 3. Oligo mapping of an RNase T1 digested sgRNA by IP-RP-LC-MS with an ACQUITY PREMIER Oligonucleotide BEH C18 300 Å, 1.7 µm 2.1 x 100 mm column as hyphenated with UV and QTof HRMS detection. (A) Full view of IP-RPLC total ion chromatogram (TIC).(B) Zoomed view of the 33 to 47 min elution window and the assignment of peak IDs including the 5’ phosphorothioate-containing oligomer, OMEG* OMEU* OMEG* CCACAGp (position 1-9) (‘r’ annotation for ribose is removed for simplicity).
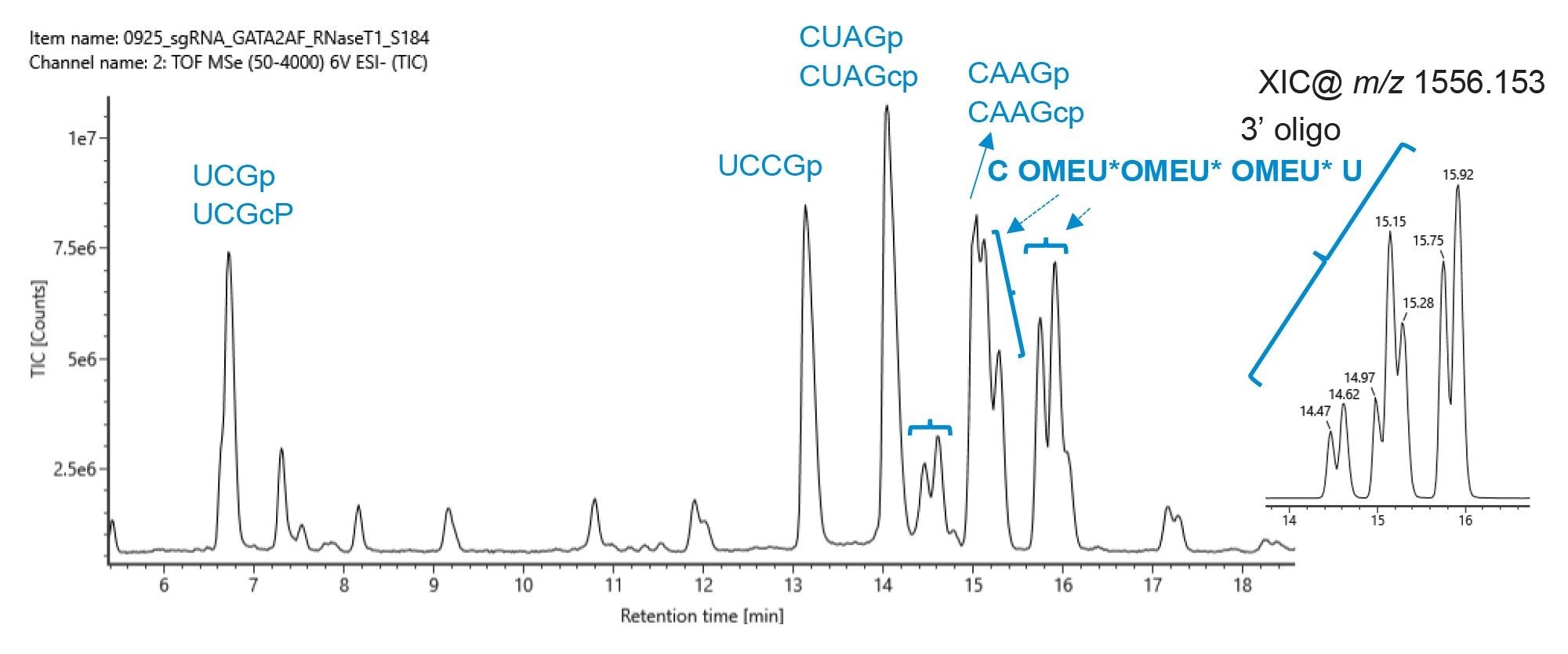 Figure 4. Oligo mapping of an RNase T1 digested sgRNA by IP-RP-LC-MS with an ACQUITY PREMIER Oligonucleotide BEH C18 300 Å, 1.7 µm 2.1 x 100 mm column as hyphenated with UV and QTof HRMS detection. Zoomed view of the 6 to 18.5 min elution window and the assignment of peak IDs. The diastereoisomers of the 3’ end of RNA with phosphorothioate-containing oligomer (C OMEU* OMEU* OMEU* U, position 96-100) are indicated by flowery brackets. The extracted ion chromatogram (XIC) for m/z 1556.1537 is shown in the inlet.
Figure 4. Oligo mapping of an RNase T1 digested sgRNA by IP-RP-LC-MS with an ACQUITY PREMIER Oligonucleotide BEH C18 300 Å, 1.7 µm 2.1 x 100 mm column as hyphenated with UV and QTof HRMS detection. Zoomed view of the 6 to 18.5 min elution window and the assignment of peak IDs. The diastereoisomers of the 3’ end of RNA with phosphorothioate-containing oligomer (C OMEU* OMEU* OMEU* U, position 96-100) are indicated by flowery brackets. The extracted ion chromatogram (XIC) for m/z 1556.1537 is shown in the inlet.
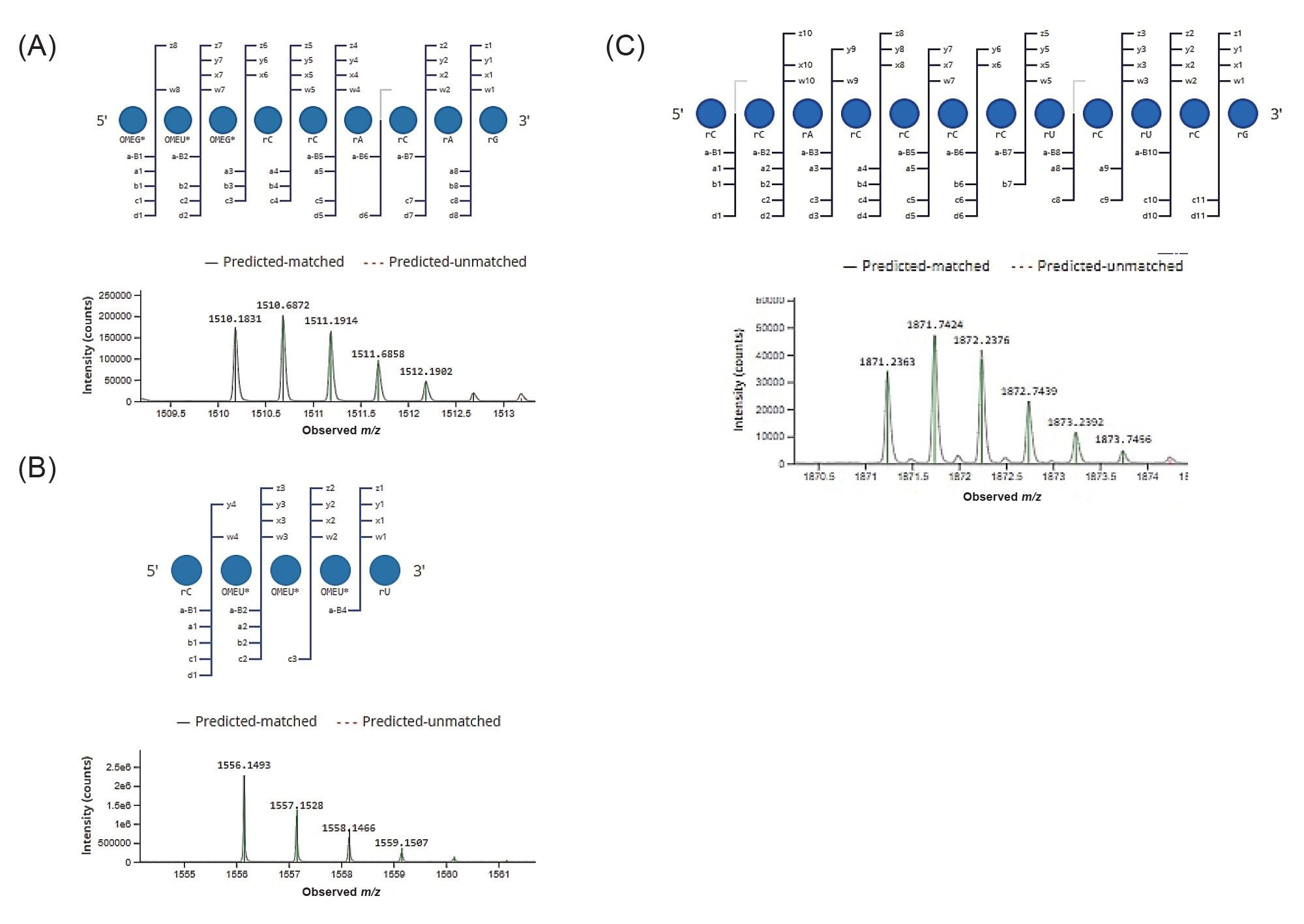 Figure 5. Example annotation of MS data for 5’ and 3’-end oligonucleotides. Dotmap representation of matched MSE fragments and low energy mass spectrum with isotope matching (highlighted in green) are shown. The 5’-oligonucleotide digestion product (position 1-9)), OMEG* OMEU* OMEG* rCrCrArCrArG (A), 3’ terminal oligonucleotide (position 96-100) rC OMEU* OMEU* OMEU* rU (B), and rCrCrArCrCrCrCrUrCrUrCrG corresponding to the position between 10–21 (C) as identified by the CONFIRM Sequence application are shown.
Figure 5. Example annotation of MS data for 5’ and 3’-end oligonucleotides. Dotmap representation of matched MSE fragments and low energy mass spectrum with isotope matching (highlighted in green) are shown. The 5’-oligonucleotide digestion product (position 1-9)), OMEG* OMEU* OMEG* rCrCrArCrArG (A), 3’ terminal oligonucleotide (position 96-100) rC OMEU* OMEU* OMEU* rU (B), and rCrCrArCrCrCrCrUrCrUrCrG corresponding to the position between 10–21 (C) as identified by the CONFIRM Sequence application are shown.
Table 1 shows the list of digestion components that were detected within the mass accuracy of precursor oligonucleotide (<7 ppm) and for which sequence-informative fragment ions could be assigned. A minimum of 80% of the expected fragment ions (at least one fragment ion for each bond) were required for a match to be made. The digest exhibited several oligonucleotides with linear as well as cyclic phosphate forms. Out of the 15 different theoretical digestion products expected from the sgRNA, three components (rUrGp, rArGp and rCrUrArGp) could have ambiguous sequence origins. The rest of the digestion components are unique and are expected to match to only one location in the sequence. Thus, 82% of the sequence can be identified by unique, sequence identifying digestion components. The coverage increases to 98% when non-unique oligonucleotides are also considered. Two G residues predicted to be released from a complete RNase T1 digestion represent the last 2% of the sgRNA here. Other nucleases and/or an experimental approach looking for missed T1 cleavages would make it possible to achieve 100% sequence coverage.
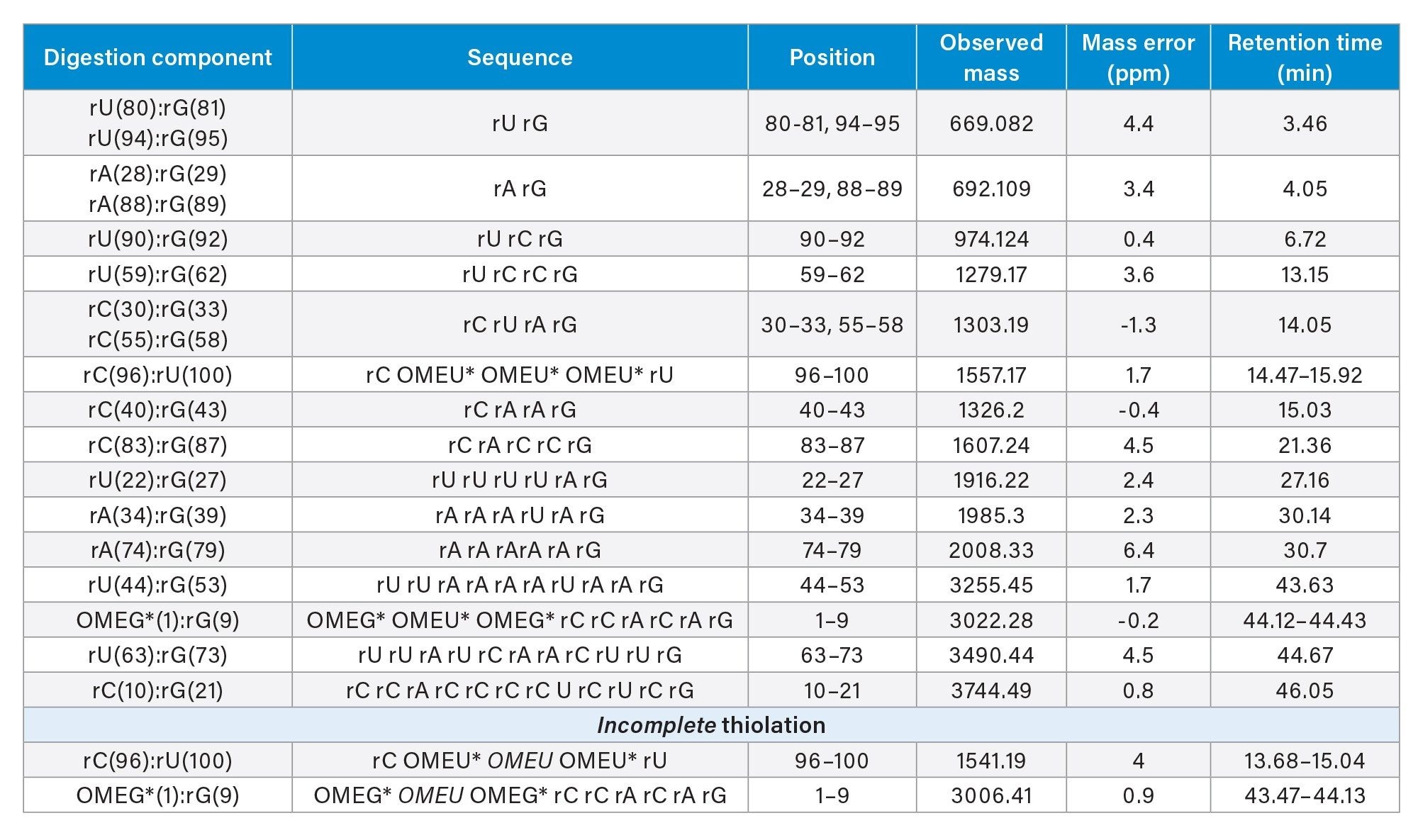 Table 1. Oligonucleotide digestion components identified using UNIFI scientific library searches and the waters_connect CONFIRM Sequence application. Oligonucleotides listed in 2nd column were identified by matching the LC-MS spectra with the predicted RNase T1 digestion products of sgRNA. IP-RP-LC-MS based detection of incomplete phosphorothiolation of RNA backbone at both 5’ and 3’ends of sgRNA sequence. * indicates a phosphorothioate linker, # indicates that a cyclic phosphate terminated species was also identified. Incomplete thiolation observed at specific positions was indicated by italic font of nucleotide.
Table 1. Oligonucleotide digestion components identified using UNIFI scientific library searches and the waters_connect CONFIRM Sequence application. Oligonucleotides listed in 2nd column were identified by matching the LC-MS spectra with the predicted RNase T1 digestion products of sgRNA. IP-RP-LC-MS based detection of incomplete phosphorothiolation of RNA backbone at both 5’ and 3’ends of sgRNA sequence. * indicates a phosphorothioate linker, # indicates that a cyclic phosphate terminated species was also identified. Incomplete thiolation observed at specific positions was indicated by italic font of nucleotide.
IP-RP-LC-MS based detection of incomplete phosphorothiolation of RNA backbone at both 5’ and 3’ends of sgRNA sequence.
The IP-RP-LC-MS data also revealed the presence of oligonucleotides with incomplete thiolation at both 5’ and 3’-regions of the sgRNA sequence, as shown in Figure 6. Significant amounts of signal (>2E4 intensity) were detected for these twice phosphorothioated (with expected 2’-O-methylation) digestion components. Extracted ion chromatograms of their precursor masses produced multipeak patterns, just like their 3 times phosphorothioated counterparts. Further, the coverage of sequence-informative fragment ions of these digestion components from the MSE fragmentation data was >93% indicating them to be confident assignments. These oligonucleotide versions were not the result of electrospray-induced oxidation as their retention times were not overlapping (Table 1). Non-overlapping profiles indicate either synthesis-related incomplete thiolation or storage-related sulfur to oxygen exchange.
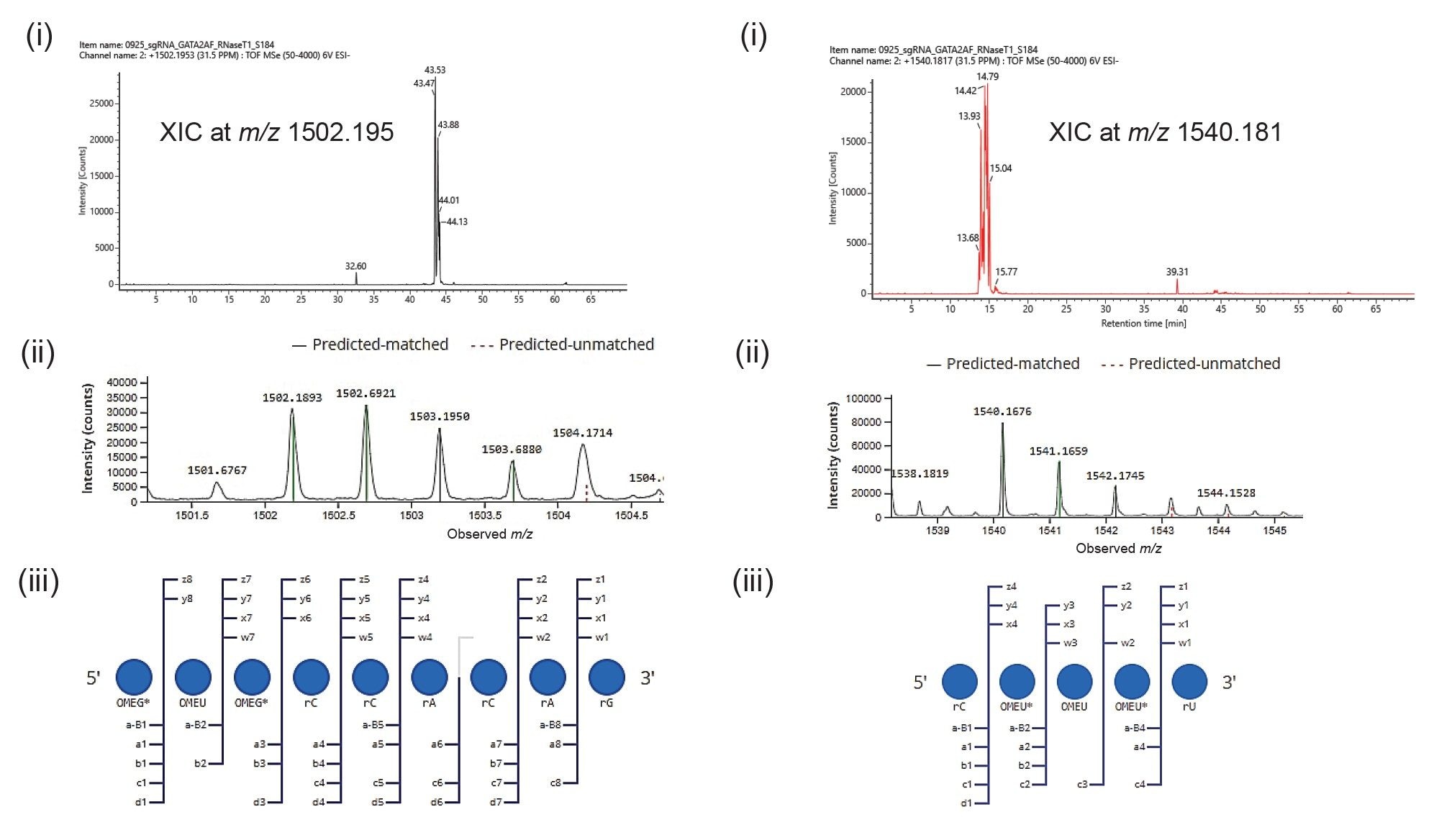 Figure 6. IP-RP-LC-MS based detection of incomplete phosphorothiolation of RNA backbone at both 5’ and 3’ends of sgRNA sequence. (A) Detection of 5’-end oligomer, OMEG* OMEU OMEG*rCrCrArCrArGp (position 1-9), with incomplete thiolation. (i) Extracted ion chromatogram (XIC) for m/z 1502.1953. (ii) Matching of the isotopic profile of the expected precursor oligonucleotide by CONFIRM sequence. (iii) Dotmap representation of the fragment ions scored for the oligonucleotide indicating the incomplete thiolation at Uridine. (B) Detection of 3’-end oligonucleotide digestion product, rC OMEU* OMEU OMEU* rU (position 96-100), with incomplete thiolation. (i) XIC for m/z 1540.1817. (ii) Matching of the isotopic profile of the expected precursor oligonucleotide by CONFIRM sequence. (iii) Dotmap representation of the matched MSE fragment ions scored for the oligonucleotide indicating the incomplete thiolation at uridine.
Figure 6. IP-RP-LC-MS based detection of incomplete phosphorothiolation of RNA backbone at both 5’ and 3’ends of sgRNA sequence. (A) Detection of 5’-end oligomer, OMEG* OMEU OMEG*rCrCrArCrArGp (position 1-9), with incomplete thiolation. (i) Extracted ion chromatogram (XIC) for m/z 1502.1953. (ii) Matching of the isotopic profile of the expected precursor oligonucleotide by CONFIRM sequence. (iii) Dotmap representation of the fragment ions scored for the oligonucleotide indicating the incomplete thiolation at Uridine. (B) Detection of 3’-end oligonucleotide digestion product, rC OMEU* OMEU OMEU* rU (position 96-100), with incomplete thiolation. (i) XIC for m/z 1540.1817. (ii) Matching of the isotopic profile of the expected precursor oligonucleotide by CONFIRM sequence. (iii) Dotmap representation of the matched MSE fragment ions scored for the oligonucleotide indicating the incomplete thiolation at uridine.
Conclusion
This application note provides an example of comprehensive sgRNA characterization that can be applied to CRISPR/Cas based genome editing reagents that are now being readied for use in medicine, diagnostics and agricultural engineering.9 Herein, we have shown a robust analytical workflow for the analysis of sgRNA at both intact and digested levels of analysis. An ACQUITY Premier BEH C18 WidePore 300 Å column used for IP-RP-LC-MS provides a reliable starting point for both types of analyses.
- Synthetic sgRNA was successfully analyzed for its integrity.
- The sgRNA intact molecular weight was deciphered by deconvolution with the waters_connect Intact Mass application.
- An RNase T1 oligo map of the sgRNA was quickly obtained without sample clean-up.
- High chromatographic resolution was achieved such that the digestion components could be readily separated and efficiently sequenced through MSE fragmentation.
- The generated T1 oligo map chromatograms were easily annotated through accurate-mass matching as facilitated by an in silico mRNA digestion calculator and the application of waters_connectTM/UNIFI scientific libraries.
- Assigned sequences for digestion components were further validated based on MSE spectra and the CONFIRM sequence application feature of the waters_connect™ platform, which gives an easy-to-interpret visual representation of fragment ion coverage.
- The phosphorothioated termini of the sgRNA could be quickly spotted in the data according to the diagnostic diastereomer separations. Moreover, incomplete thiolation of these species could be distinguished and detected.
References
- Barrangou, R., et al., CRISPR provides acquired resistance against viruses in prokaryotes. Science, 2007. 315(5819): p. 1709–12.
- Deltcheva, E., et al., CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature, 2011. 471(7340): p. 602–7.
- Jinek, M., et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 2012. 337(6096): p. 816–21.
- Shao, M., T.R. Xu, and C.S. Chen, The Big Bang of Genome editing technology: development and application of the CRISPR/Cas9 system in disease animal models. Dongwuxue Yanjiu, 2016. 37(4): p. 191–204.
- Mohr, S.E., et al., CRISPR guide RNA design for research applications. Febs j, 2016. 283(17): p. 3232–8.
- Pennisi, E., The CRISPR craze. Science, 2013. 341(6148): p. 833–6.
- Charpentier, E., A. Elsholz, and A. Marchfelder, CRISPR-Cas: more than ten years and still full of mysteries. RNA Biology, 2019. 16(4): p. 377–379.
- Maissa M. Gaye, J.F., Johannes P.C. Vissers, Ian Reah, Chris Knowles, and Matthew A. Lauber, Synthetic mRNA Oligo-Mapping Using Ion-Pairing Liquid Chromatography and Mass Spectrometry. Waters Application Note, 720007669, 2022.
- Asmamaw, M. and B. Zawdie, Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biologics, 2021. 15: p. 353–361.
720007897, March 2023