Total Workflow for the Sensitive Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Fish, Meat, Edible Offal, and Eggs
Abstract
Environmental release and contamination of per- and polyfluorinated alkyl substances (PFAS) has resulted in contamination of a variety of food sources. Complex food commodities such as fish, meat, edible offal, and eggs require a comprehensive sample extraction and clean up. To accommodate these types of samples, an alkaline digestion and extraction was implemented followed by Weak Anion Exchange (WAX) SPE to produce a suitable sample for analysis. High sensitivity LC-MS/MS analysis was performed on an ACQUITY UPLC I–Class PLUS coupled to Xevo TQ-XS. The method was evaluated in six different commodity types including salmon, tilapia, ground beef, beef liver, beef kidney, and chicken eggs. This approach proved to be accurate, sensitive, and robust for a range of 30 PFAS compounds of varying chemistry classes to match the challenging concentrations published in reports by the European Food Safety Authority (EFSA) and the US Food and Drug Administration (FDA).
Benefits
- A single extraction method that can be utilized for a large suite of PFAS from a variety of food matrices
- Sensitive analysis on the Xevo TQ-XS to detect PFAS at sub-ng/g levels to match the challenging concentrations published in reports by EFSA and FDA
- Confidence in results with the utilization of the PFAS Kit for LC modification minimize possible system and solvent contaminants to assure accurate results
Introduction
Rising concerns about the long-term impacts of human exposure to per- and polyfluoroalkyl substances (PFAS) have propelled the scope of PFAS analysis from just environmental matrices into the field of food analysis as well. Over the last decade, cases of PFAS contamination being found in foods such as, but not limited to, eggs, milk, chocolate cake, and fast food have become more prominent in the media. To protect the public and understand dietary exposure, analytical methods for the analysis of a large variety of food products are required.
In response, many agencies around the globe have been studying the impact of PFAS in food. The most recent report published in 2020 by the European Food Safety Authority (EFSA) have identified fish, meat, fruit/fruit products, and eggs/egg products to contribute most to human PFAS exposure through diet during the study period of 2007 to 2018.1 From this study, EFSA set a recommended tolerable weekly intake (TWI) of 4.4 ng per kg of body weight for a total of 4 PFAS: PFOA, PFNA, PFHxS, and PFOS.
Among food derived from animal origin, the EFSA study found the mean concentration of PFAS identified varied by sample type. PFOA, PFNA, PFHxS, and PFOS were found in livestock and wild game meats in the range of 0.02–1.59 ng/g, in farm raised edible offal in the range of 0.087–1.18 ng/g, in salmon and trout in the range of 0.003–0.83 ng/g and in eggs and egg products in the range of 0.06–0.35 ng/g. The median detected concentration in all sample types considered was 0.32 ng/g. In addition to the EFSA study, the FDA monitor contaminants, including PFAS, in highly consumed foods in their Total Diet Study (TDS).2 In the most recent study from 2019, the TDS reported a finding of 0.087 ng/g PFOS in tilapia. These studies and reports provide evidence for the sensitivity required in a method suitable to analyze these types of samples.
A strategy for the extraction of PFAS from edible produce was discussed in a previous application note,3 therefore the focus of this study will be meat, edible offal, fish, and eggs. These food commodities were separated due to differences in sample preparation needs. Produce is a simpler matrix to extract and utilized a QuEChERS extraction and dispersive SPE protocol. Animal based samples add complexity to sample preparation needs due to the presence of proteins and fats that can bind PFAS. There are a variety of approaches available for the extraction of PFAS from meat, fish, and similar matrices. One of the most effective methods is to perform an alkaline extraction (typically sodium hydroxide in methanol or acetonitrile) of the sample followed by solid phase extraction (SPE) clean-up.4,5 This is the approach this study has taken to evaluate the extraction of a suite of 30 PFAS from six different food matrices: salmon, tilapia, ground beef, beef liver, beef kidney, and egg. Weak Anion Exchange (WAX) chemistry was utilized for the SPE clean-up. Analysis was performed on an ACQUITY UPLC I–Class PLUS modified with PFAS Kit and coupled to a Xevo TQ-XS to fit the highly sensitive analysis requirements.
Experimental
Sample Preparation
Samples of frozen salmon, frozen tilapia, ground beef, beef liver, beef kidney, and whole chicken eggs were purchased from local grocery stores. Fish and meat were homogenized using a Ninja kitchen blender. These samples were stored in a freezer (-20 °C) and thawed in a refrigerator (4 °C) overnight prior to extraction. Eggs were prepared fresh. After removing from the shell, the egg white and yolk were mixed before subsampling. All standards were obtained from Wellington Laboratories. The method contained a total of 30 PFAS including the following compounds: Carboxylates: C4–C14; Sulfonates: C4–C10; Ethers: GenX, ADONA, 9Cl-PF3ONS, 11Cl-PF3OUdS; Precursors: FBSA, FHxSA, FOSA, NMeFOSAA, NEtFOSAA, 4:2 FTS, 6:2 FTS, 8:2 FTS.
Prior to extraction, a 2 g sample was weighed into a 50 mL centrifuge tube and spiked with 1 ng/g extraction standard (MPFAC-24ES and M3-HFPODA). 10 mL methanol containing 0.02 M sodium hydroxide was added to each sample. Samples were shaken for 1 hour using a SPEX Sample Prep Geno/Grinder at 500 RPM. After shaking, samples were centrifuged for 10 minutes at 4000 RPM at room temperature. Following extraction, 0.5 mL of supernatant was diluted in 14.5 mL water in preparation for solid phase extraction (SPE) using Oasis WAX for PFAS, 6 cc, 150 mg Cartridges (p/n: 186009345). The full SPE procedure is detailed in steps 2–5 of Figure 1.
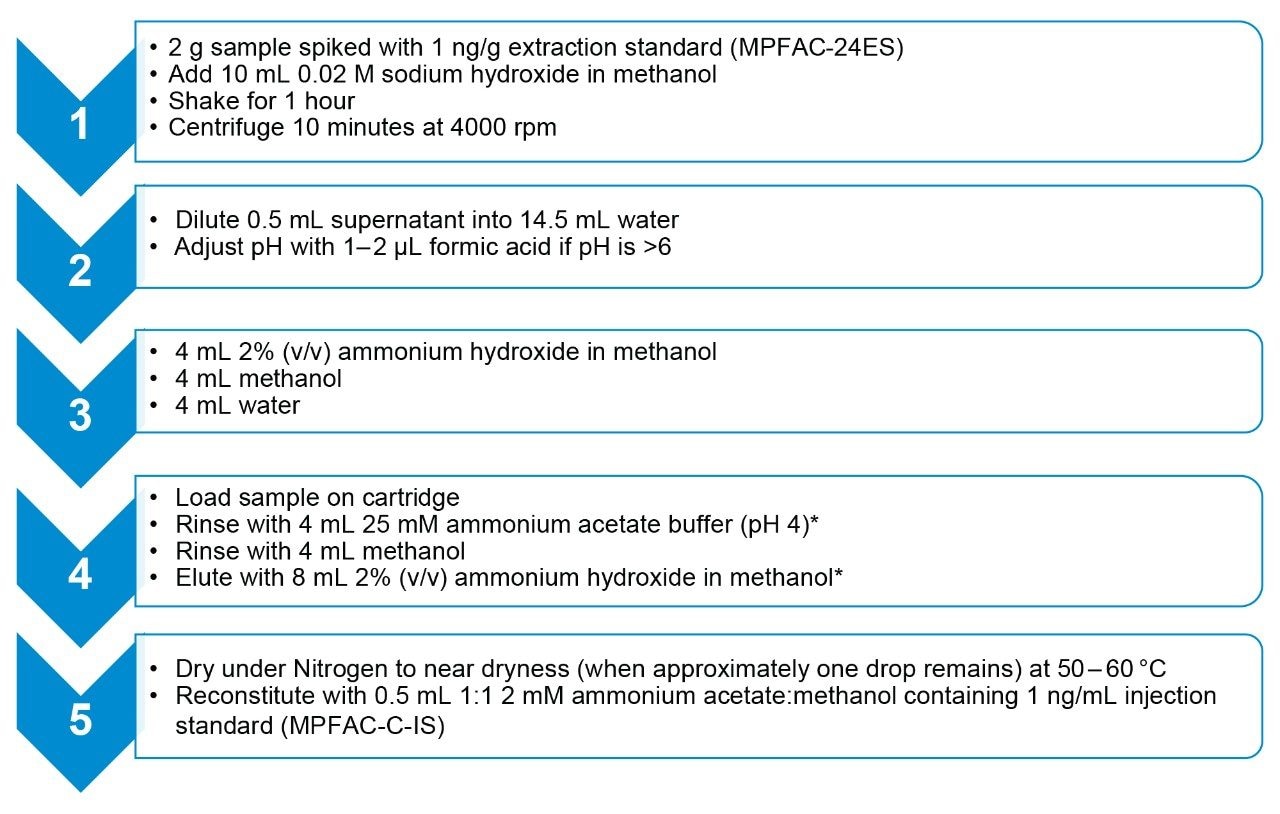 Figure 1. Procedure for SPE clean-up of extracts. Steps denoted with (*) indicate the solvent used in this step should be used to rinse the sample tube prior to addition to the SPE cartridge.
Figure 1. Procedure for SPE clean-up of extracts. Steps denoted with (*) indicate the solvent used in this step should be used to rinse the sample tube prior to addition to the SPE cartridge.
A solvent calibration curve in the range of 0.01–5 ng/mL (equivalent to 0.05–25 ng/g) was used for sample analysis. For limit of detection (LOD) and limit of quantitation (LOQ) assessment, matrix post spiked (or matrix matched) calibration curves in the range of 0.001–5 ng/mL were used. Isotope labelled extraction (MPFAC-24ES and M3-HFPODA) and injection standards (MPFAC-24ES) were used during extraction and analysis to perform isotope dilution calculations. The extraction standard was spiked in the samples prior to sample preparation and used to correct the native compounds for recovery and matrix effects. The injection standard was added to the sample after clean-up when the sample was reconstituted and used to correct the extraction standards for reconstitution variations, matrix effects, and injection variation. With the presence of the extraction and injection standards, matrix matching was not necessary for routine sample analysis, but is an option in place of performing the isotope dilution method.
LC Conditions
|
LC system: |
ACQUITY UPLC I–Class PLUS with PFAS Analysis Kit |
|
Vials: |
Polypropylene autosampler vial with polyethylene cap (p/n: 186005230) |
|
Column(s): |
ACQUITY UPLC BEH C18 2.1x 100 mm, 1.7 um (p/n: 186002352) with ACQUITY Column In Line Filter (p/n: 205000343) |
|
Column temp.: |
35 °C |
|
Sample temp.: |
4 °C |
|
Injection volume: |
10 µL |
|
Flow rate: |
0.3 mL/min |
|
Mobile phase A: |
Water + 2 mM Ammonium acetate |
|
Mobile phase B: |
Methanol + 2 mM Ammonium acetate |
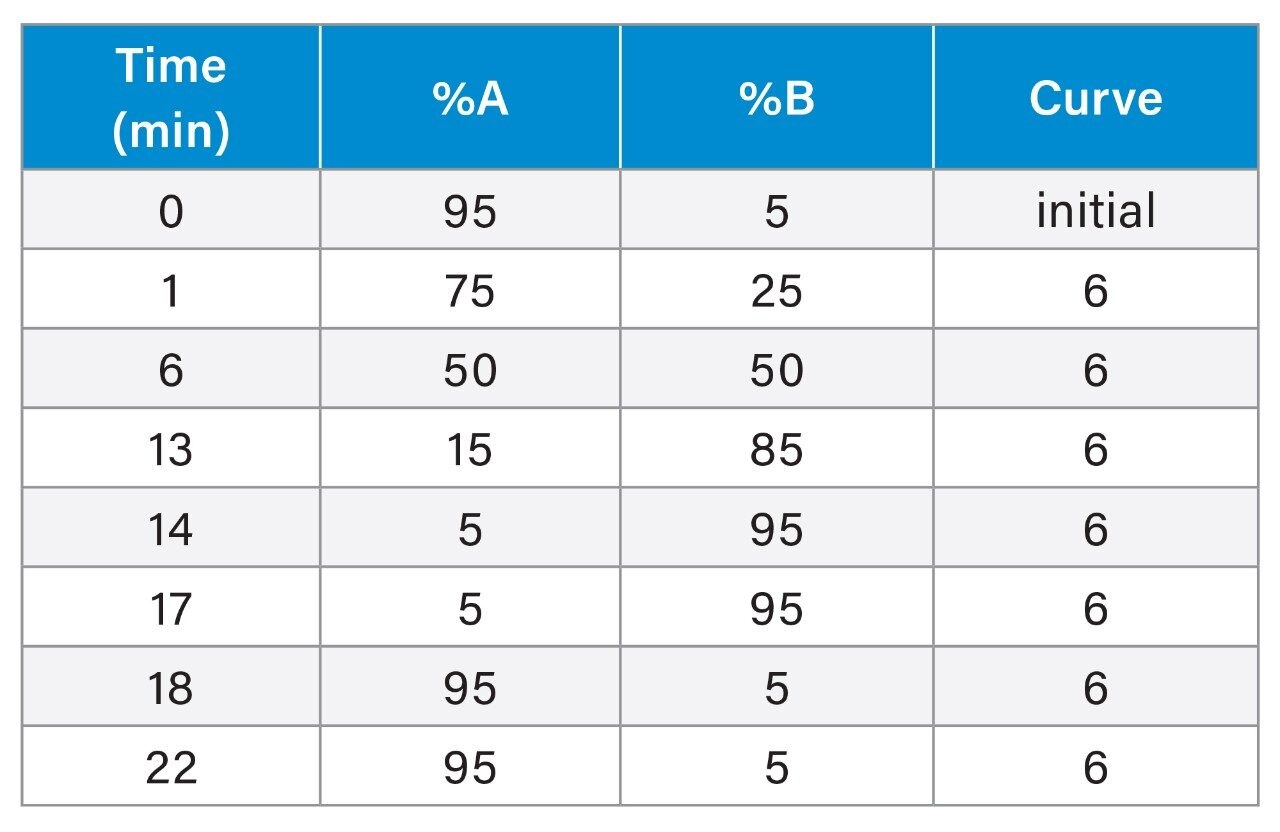
Gradient Table
MS Conditions
|
MS system: |
Xevo TQ-XS |
|
Ionization mode: |
ESI- |
|
Source temp.: |
100 °C |
|
Capillary voltage: |
0.50 kV |
|
Desolvation temp.: |
350 °C |
|
Desolvation flow: |
900 L/hr |
|
Cone flow: |
150 L/hr |
|
MRM method: |
See Appendix for Full MRM Method details |
Data Management
|
Chromatography software: |
MassLynx v4.2 |
|
MS software: |
MassLynx v4.2 |
|
Informatics: |
MassLynx v4.2 with TargetLynx v4.2 |
Results and Discussion
The method was evaluated using five replicates of each commodity spiked at 3 concentration levels; 0.1 ng/g, 1.0 ng/g, and 5 ng/g. Example chromatograms of the extracted quantitation ion trace of each PFAS spiked into salmon prior to extraction at the lowest level of 0.1 ng/g can be seen in Figure 2. Limit of detection and quantitation (LOD and LOQ) were also determined for all samples except for egg and are reported in the Appendix Table 2. LOD and LOQ values were calculated from matrix blank extracts of each sample post spiked with PFAS standards. Calculations were performed in TargetLynx using the criteria of S:N of 3 for LOD and 10 for LOQ.
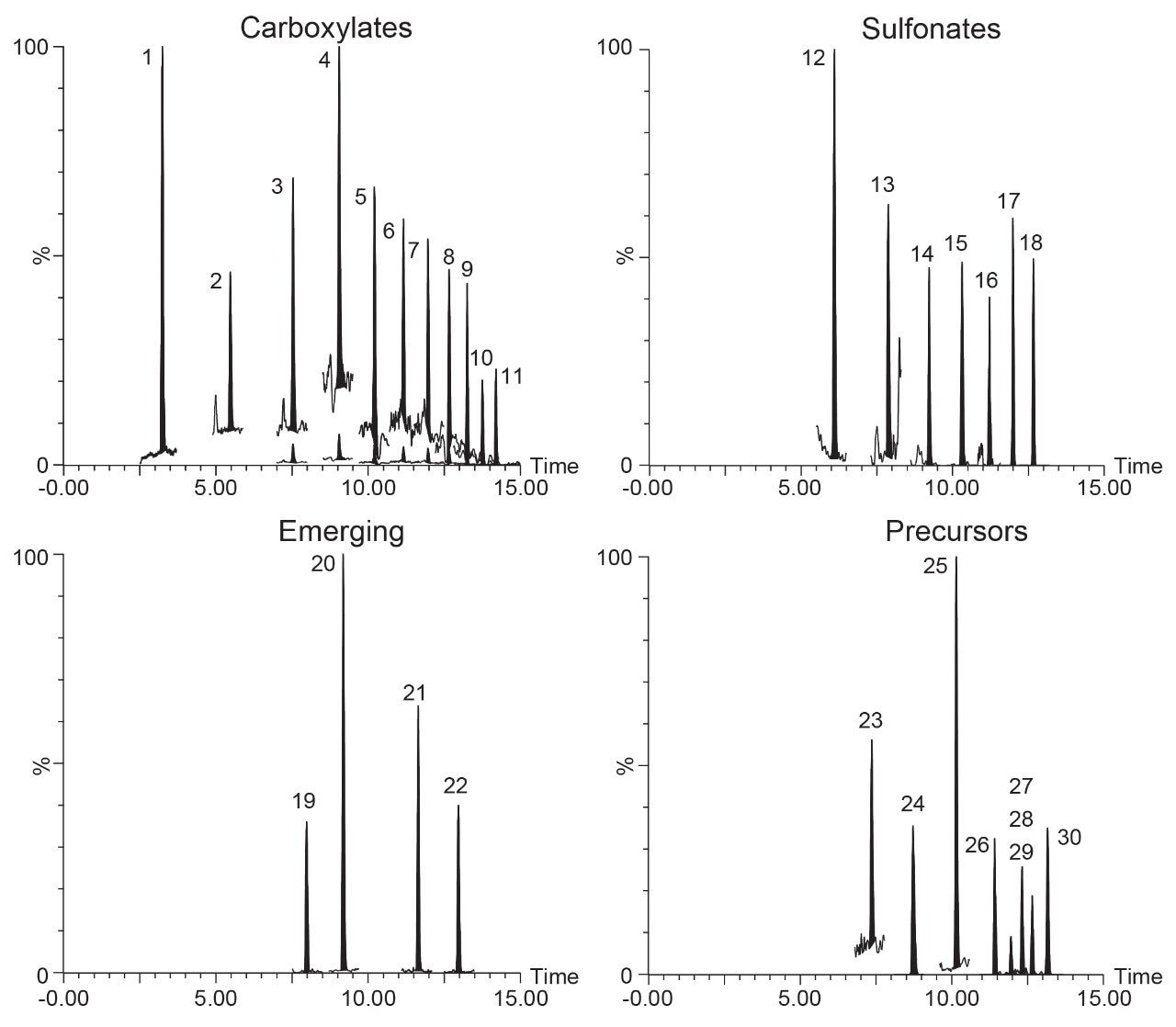 Figure 2. Extracted ion chromatograms of the quantitation ion for each PFAS in salmon spiked at 0.1 ng/g (Peak 23 shown at 1.0 ng/g). Peak assignments are listed in the Appendix. Note: Peaks 3–11 shown zoomed in to display on same view as Peaks 1 and 2.
Figure 2. Extracted ion chromatograms of the quantitation ion for each PFAS in salmon spiked at 0.1 ng/g (Peak 23 shown at 1.0 ng/g). Peak assignments are listed in the Appendix. Note: Peaks 3–11 shown zoomed in to display on same view as Peaks 1 and 2.
A well-known matrix interferent compound in meat and meat-like samples is taurodeoxycholic acid (TDCA). This is a group of isomers that have a nominal molecular mass of 499.7 amu and share the same 499 > 80 MRM transition commonly used as the PFOS quantitative transition. TDCA was detected in beef liver and chicken eggs during this study, but using 499 > 91 MRM as the quantitative transition was specific to PFOS. Figure 3A demonstrates the use of the 499 > 91 MRM transition for PFOS in liver and eggs as well as the comparison to ground beef, which did not contain this interferent (fish and kidney did not either). In addition to the alternate transition, Figure 3B demonstrates the presence of TDCA did not cause any matrix suppression or enhancement in liver (due to the chromatographic resolution and specificity of the 499 > 91 transition) as demonstrated by the graphed peak areas of the injection standard M-PFOS during a continuous sample analysis including salmon, tilapia, ground beef, liver, and kidney. Ground beef did appear to have another source of matrix suppression, as demonstrated in Figure 3B, but this was not from TDCA and not an isobaric interferent that could be detected in the M-PFOS MRM transition. Even with the presence of matrix effects demonstrated in beef, utilization of the injection and extraction internal standards allowed for accurate and reproducible quantitation, as demonstrated and discussed later in Table 1.
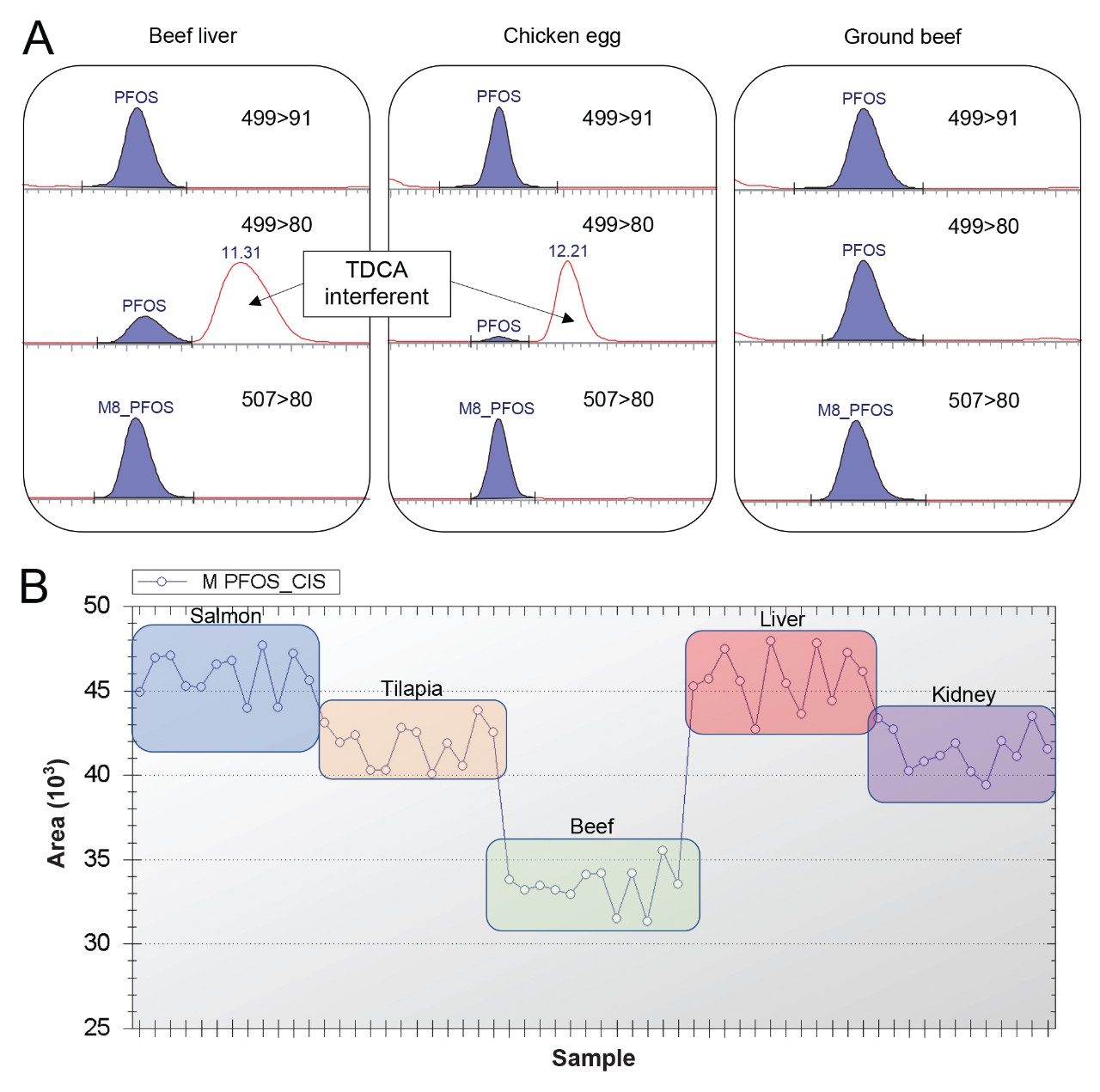 Figure 3. (A) chromatograms of PFOS MRM transitions 499 > 91 and 499 > 80 and M8-PFOS in beef liver, chicken egg, and ground beef showing taurodeoxycholic acid (TDCA) as an interferent in 499 > 80 transition in liver and egg. (B) M-PFOS injection standard peak area (y-axis) over 12 sample injections of each sample matrix (x-axis): salmon, tilapia, beef, liver, and kidney.
Figure 3. (A) chromatograms of PFOS MRM transitions 499 > 91 and 499 > 80 and M8-PFOS in beef liver, chicken egg, and ground beef showing taurodeoxycholic acid (TDCA) as an interferent in 499 > 80 transition in liver and egg. (B) M-PFOS injection standard peak area (y-axis) over 12 sample injections of each sample matrix (x-axis): salmon, tilapia, beef, liver, and kidney.
The isotope labelled extraction standards were used to evaluate method recovery due to lack of a truly blank matrix and issues with extraction solvent contamination. Recovery values are shown in Figure 4, with standard deviation for n=15 extracts. There currently are no official guidelines or standards for PFAS in food to compare the recovery results to. The FDA has a set of general guidelines regarding validation of chemical methods in various matrices, including food, that can be referenced for recovery guidelines.6 This document allows for a recovery range of 40–120% at a concentration of 1 ng/g. The neutral sulfonamides are not recovered using the WAX SPE protocol as they are lost to waste during the methanol wash step required to remove matrix, resulting in the low recoveries in Figure 4 for 13C8-FOSA (perfluorooctane sulfonamide). Alternate SPE using Oasis HLB can be utilized if recovery of the sulfonamides is required but is not suitable for the full range of PFAS compounds covered in this study. Besides the sulfonamides, the long chain carboxylates were difficult to recover from egg, salmon, and tilapia, resulting in recoveries below the FDA guidelines of 40%. Additionally, NEtFOSAA had recovery of 30% in tilapia. Besides these problematic compounds, the remaining PFAS recoveries were within the FDA guidelines.
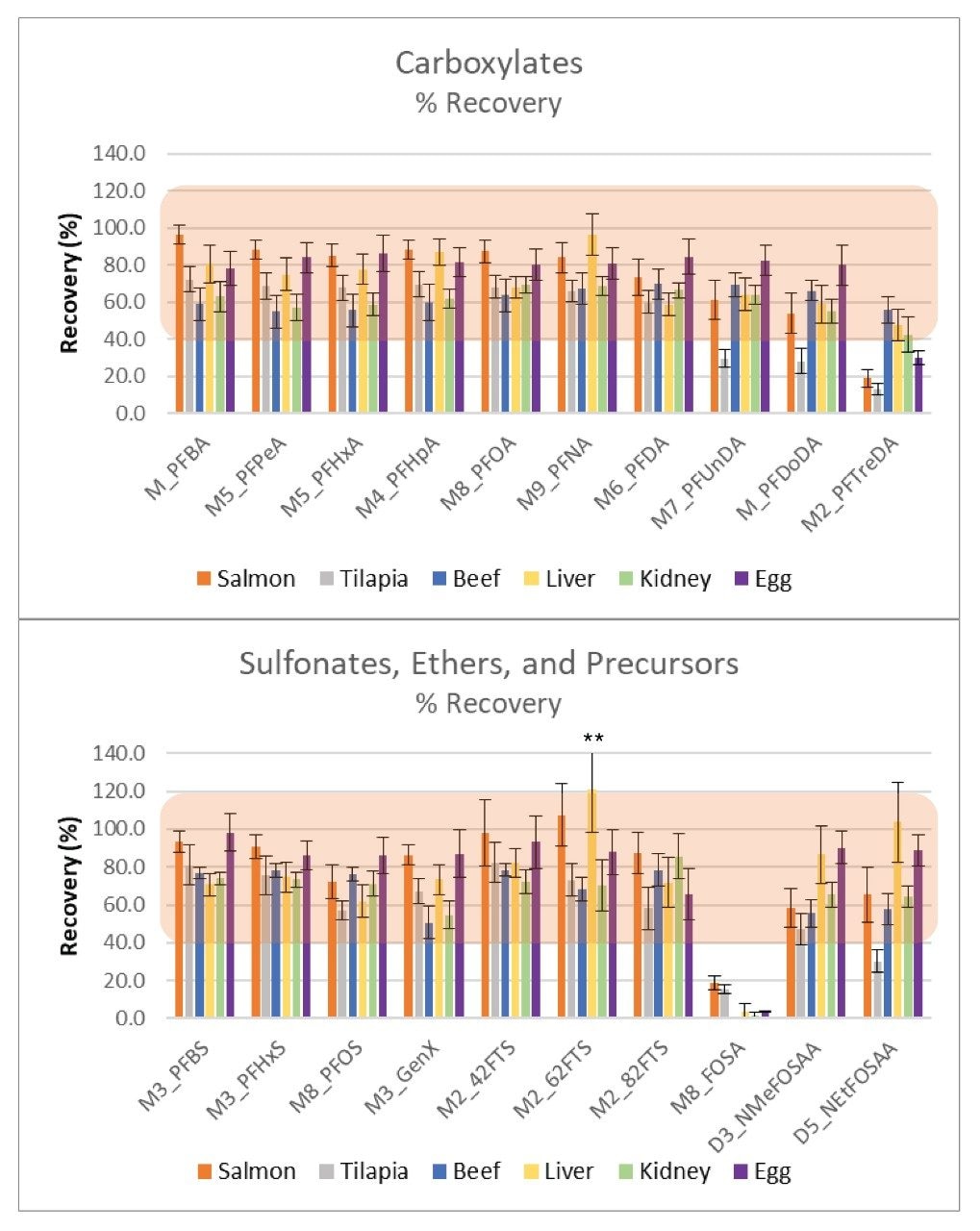 Figure 4. Percent recovery in each matrix evaluated, n=15 for all matrices except egg where n=5. **143% standard deviation shown off scale. Orange highlight demonstrates the FDA guidelines for recovery at 1 ng/g (40–120%).
Figure 4. Percent recovery in each matrix evaluated, n=15 for all matrices except egg where n=5. **143% standard deviation shown off scale. Orange highlight demonstrates the FDA guidelines for recovery at 1 ng/g (40–120%).
Accuracy of the method was evaluated by percent bias of the comparison of calculated concentration to the expected concentration for each PFAS at the three different spike levels. Table 1 lists the mean calculated concentrations (using the isotope dilution method) for each type of sample as well as the percent bias, or accuracy. For the 1.0 and 5.0 ng/g spike levels, as well as the 0.1 ng/g spike level for kidney, liver, tilapia, and beef, mean concentrations were within 17% of the expected value. Liver and salmon were within 30% of the expected value for the 0.1 ng/g level. The %RSD values for five replicates of each concentration is also shown in Table 1. RSDs were below 20% for all samples except for the 0.1 ng/g spike level of liver and salmon having RSDs of 21.9 and 28.4, respectively. The bias and RSD values demonstrate both precision and reproducibility of the method.
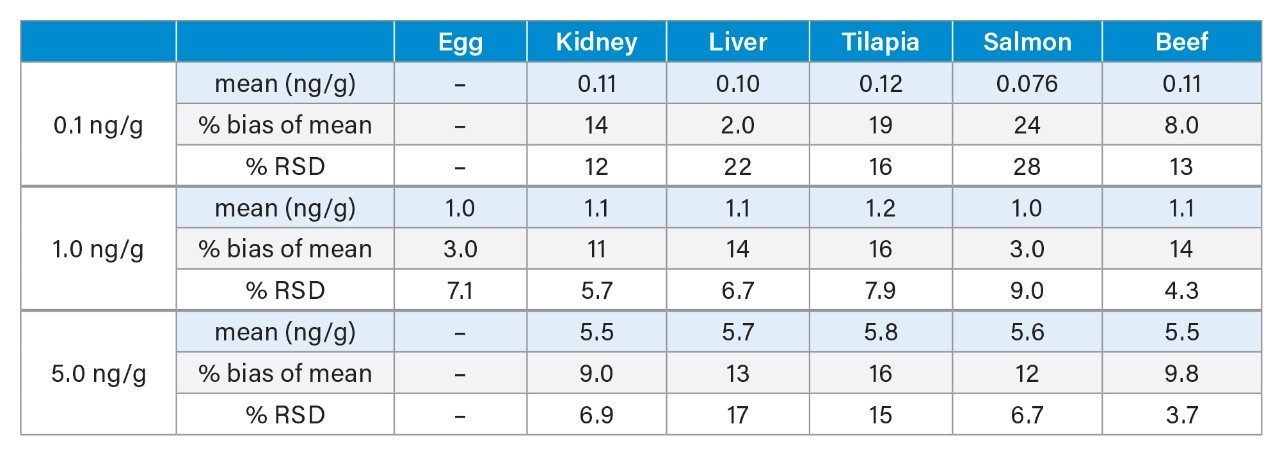 Table 1. Mean calculated concentration, %bias of mean, and %RSD (n=5) for each spike level in egg, kidney, liver, tilapia, salmon, and beef.
Table 1. Mean calculated concentration, %bias of mean, and %RSD (n=5) for each spike level in egg, kidney, liver, tilapia, salmon, and beef.
In addition, NIST standard reference material 1947, Lake Michigan Fish Tissue, was extracted and analyzed alongside the spiked samples to gauge the accuracy of the method. This reference material reports NIST determined concentrations for four PFAS (PFNA, PFDA, PFUnDA, PFTriDA), but does not indicate uncertainty values due to lack of data. During analysis, n=8 replicates of NIST 1947 were extracted and analyzed and the comparison data is reported in Figure 5. While the uncertainty values aren’t available for the NIST SRM, the experimental results are not significantly different to the NIST values, further demonstrating method accuracy.
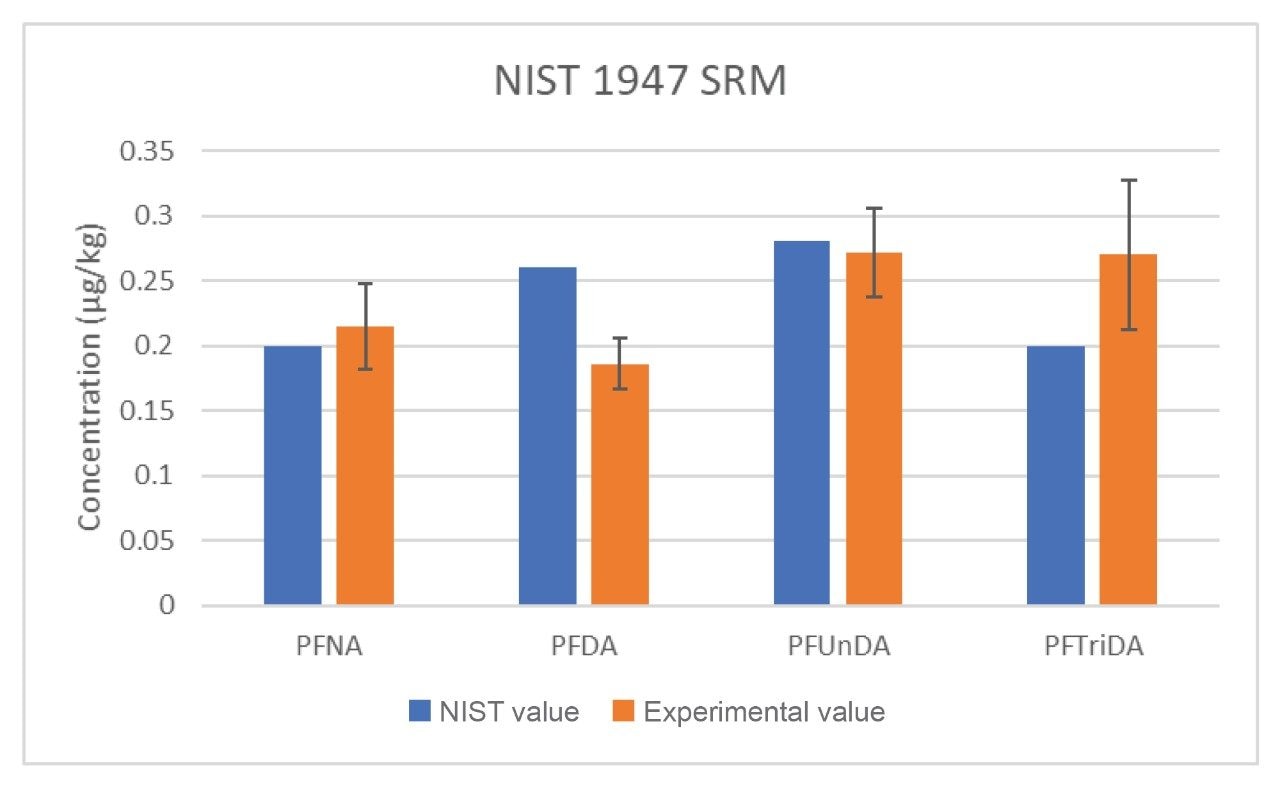 Figure 5. Comparison of NIST reported values to experimental values of four PFAS in NIST 1947 SRM, Lake Michigan Fish Tissue. n=8 for experimental values.
Figure 5. Comparison of NIST reported values to experimental values of four PFAS in NIST 1947 SRM, Lake Michigan Fish Tissue. n=8 for experimental values.
Finally, there were detectable amounts of PFAS in the chicken egg and beef liver samples used in this study that were confidently identified and quantified (Figure 6). Beef liver contained 0.76 ng/g PFOS (0.52 ng/g linear, 0.24 ng/g branched), whereas chicken eggs contained PFPeA, PFHxA, PFHpA, and PFOA in amounts of 0.18, 0.25, 0.29 and 0.13 ng/g, respectively.
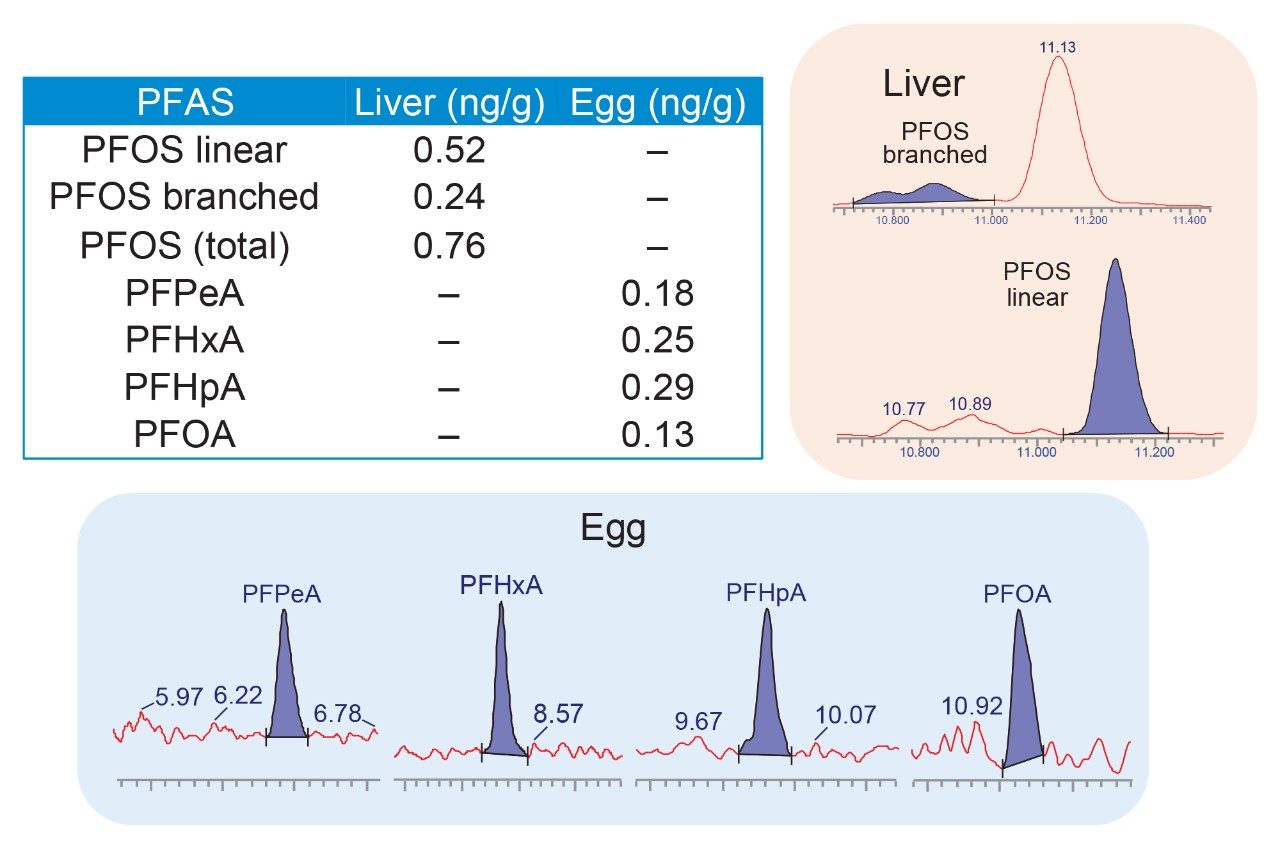 Figure 6. PFAS detected in samples of beef liver and egg purchased in local grocery stores.
Figure 6. PFAS detected in samples of beef liver and egg purchased in local grocery stores.
Conclusion
Rising concerns about the long-term impacts of human exposure to per- and polyfluoroalkyl substances (PFAS) have propelled the scope of PFAS analysis from just environmental matrices into the field of food analysis. To protect the public and understand dietary exposure, analytical methods for the analysis of a large variety of food products are required. EFSA has identified food from animal origin to contribute the most to human exposure to PFAS. In response, an alkaline digestion extraction followed by WAX SPE clean-up was successfully implemented to extract PFAS from challenging food samples of meat, fish, edible offal, and eggs. Sensitive and accurate analysis of extracts was performed using the Xevo TQ-XS allowing for detection and quantitation limits in the sub-ng/g range required to meet the maximum dietary levels of PFAS recommended by EFSA. Utilization of the PFAS Kit for LC modification also provides confidence in results by minimizing system and solvent contaminants. Recoveries were within FDA criteria for all compounds except the C13 and C14 carboxylates. Utilization of the isotope dilution method does take the recovery into account and allows for accurate correction of recovery during calculation of PFAS concentration in samples. Five PFAS were detected in two different food samples purchased from local grocery stores. This comprehensive method allows for high confidence in results of PFAS in complex food matrices to allow for better monitoring and understanding of the environmental impact of PFAS on our food sources.
References
- Schrenk D, Bignami M, et al. EFSA Panel on Contaminants in the Food Chain (EFSA CONTAM Panel), Risk to Human Health Related to the Presence of Perfluoroalkyl Substances in Food. EFS2. 2020;18(9).
- Food and Drug Administration. Analytical Results of Testing Food for Pfas From Environmental Contamination. June 2021 [cited 2021 December 15]. Available from: https://www.fda.gov/food/chemical-contaminants-food/analytical-results-testing-food-pfas-environmental-contamination.
- Organtini K, Hird S, Adams S. QuEChERS Extraction of Per- and Polyfluoroalkyl Substances (PFAS) from Edible Produce with Sensitive Analysis on Xevo TQ-XS. Waters Application Note, 720007333, August 2021.
- Sadia M, Yeung LWY, Fiedler H. Trace Level Analyses of Selected Perfluoroalkyl Acids in Food: Method Development and Data Generation. Environmental Pollution. 2020;263.
- Delinsky AD, Strynar MJ, Nakayama SF, Varns JL, Ye X, McCann PJ, Lindstrom AB. Determination of Ten Perfluorinated Compounds in Bluegill Sunfish (Lepomis Macrochirus) Fillets. Environmental Research. 2009;109:975–984.
- Food and Drug Administration Foods Program. Guidelines for the Validation of Chemical Methods in Food, Feed, Cosmetics, and Veterinary Products, 3rd edition. October 2019.
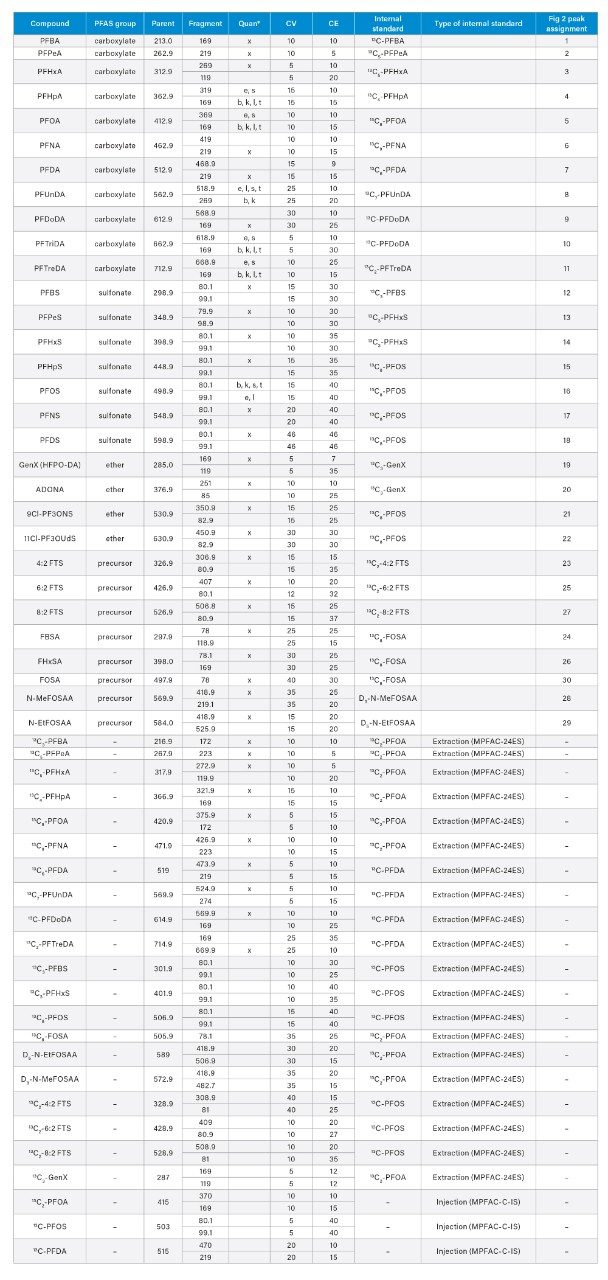 Appendix Table 1. MS Method conditions for PFAS included in method. (*) x indicates MRM used as quan ion for all matrices. The following letters indicate the MRM was used as the quan ion for the type of designated matrix (s) salmon, (t) tilapia, (b) ground beef (l) liver (k) kidney, and (e) egg.
Appendix Table 1. MS Method conditions for PFAS included in method. (*) x indicates MRM used as quan ion for all matrices. The following letters indicate the MRM was used as the quan ion for the type of designated matrix (s) salmon, (t) tilapia, (b) ground beef (l) liver (k) kidney, and (e) egg.
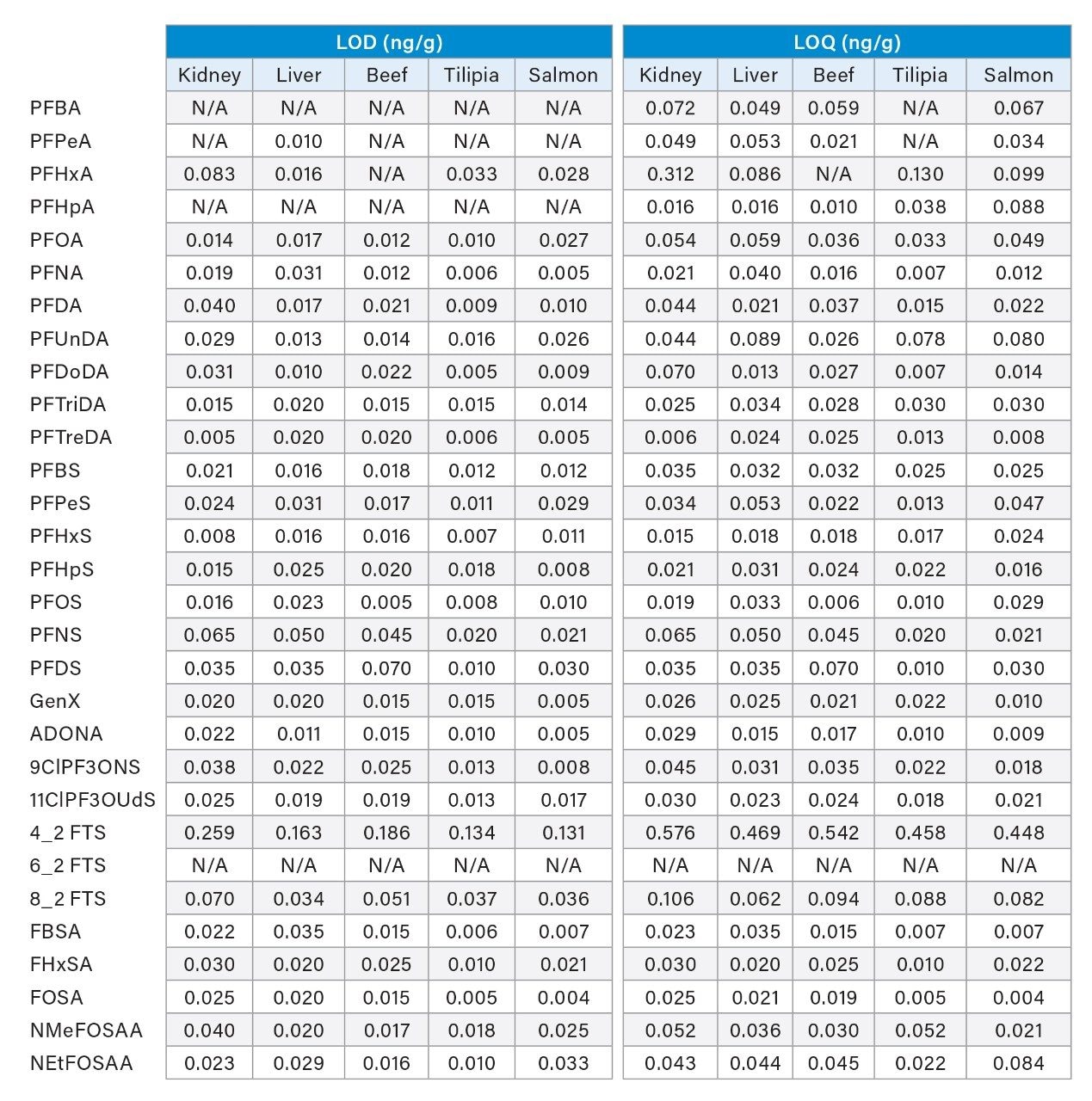 Appendix Table 2. LOD and and LOQ for each matrix reported in ng/g.
Appendix Table 2. LOD and and LOQ for each matrix reported in ng/g.
720007482, January 2022