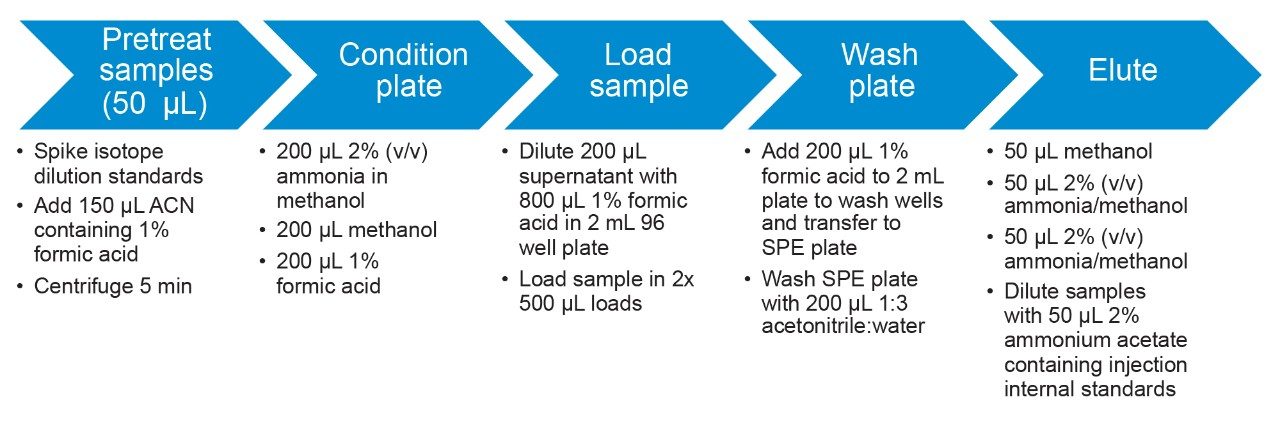
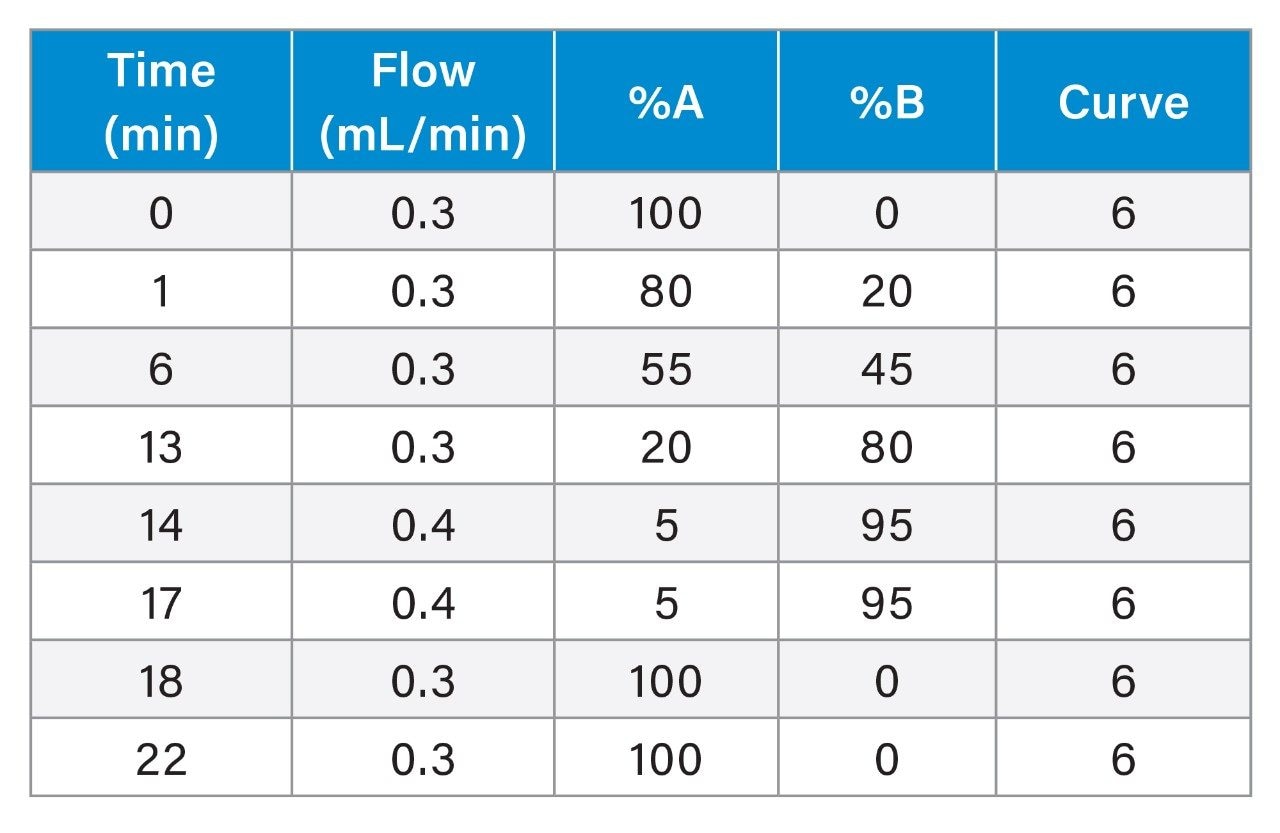
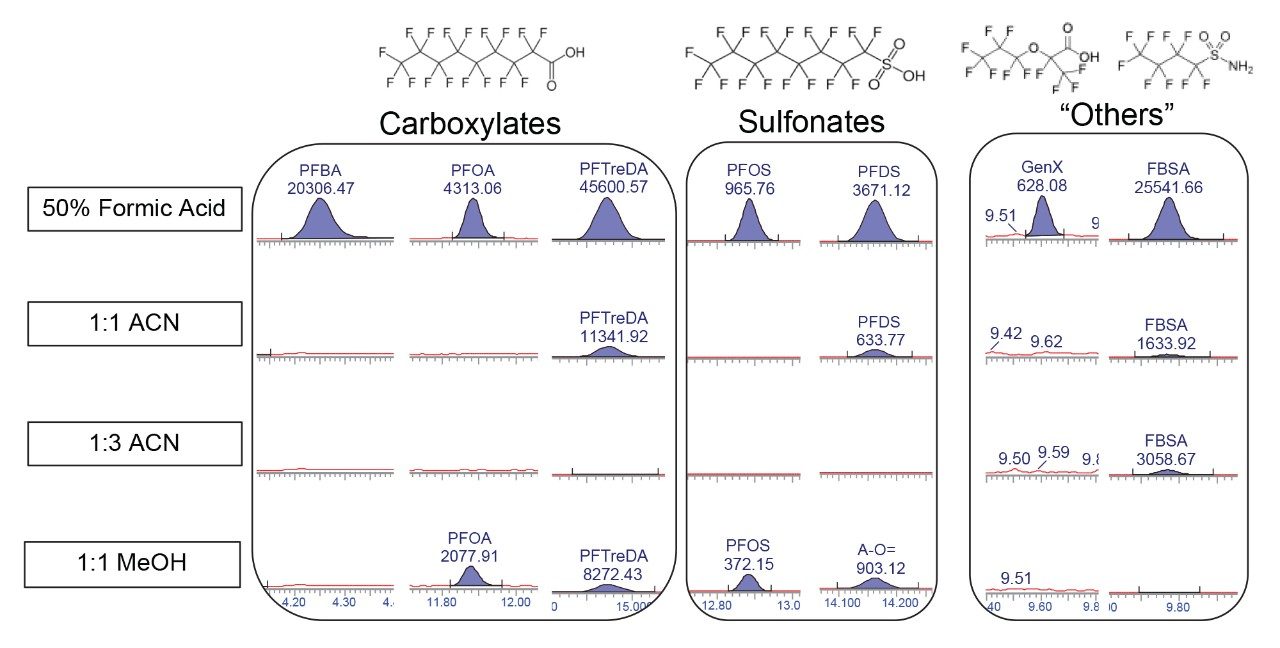
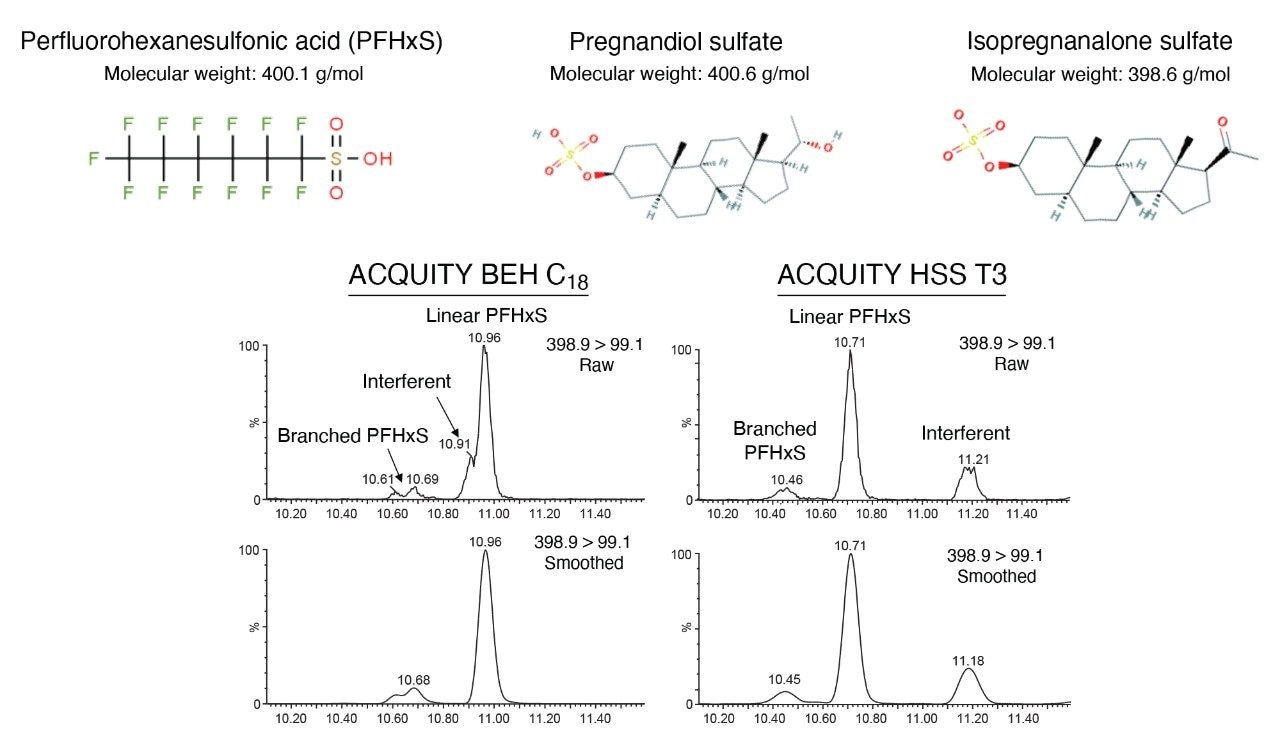
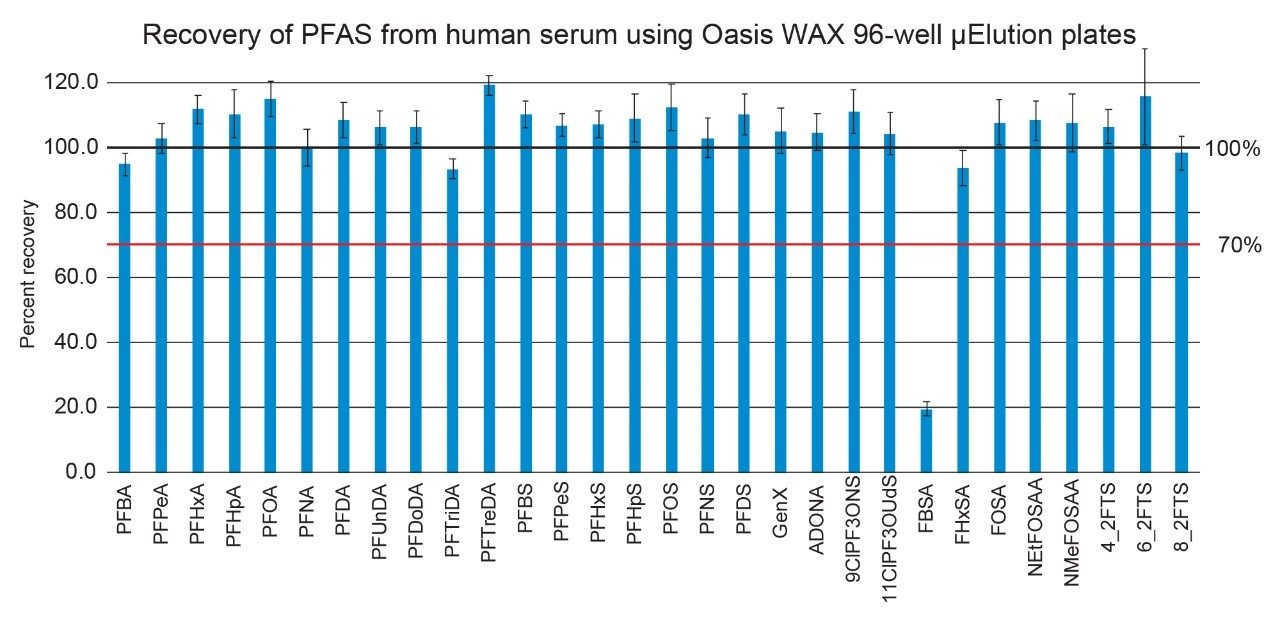
With the exception of the C4 sulfonamide (FBSA), recoveries
were within an acceptable range of 85 to 120%. Recovery of FBSA was
approximately 20% and is due to this compound being a low molecular weight,
neutral PFAS compound that may not be best suited to a multiresidue extraction.
This compound also did not have a suitable internal standard available to aid
in isotope dilution correction. Although FBSA recovery was low, it still was
detectable allowing for identification, if present, in a sample. The SPE
procedure could be slightly altered if increased recovery of this compound was
crucial. For example, the second SPE plate wash using 1:3 acetonitrile:water
could be eliminated to recover more FBSA. Alternately, if neutral compounds
were the only PFAS of interest, a reverse phase chemistry, such as Oasis HLB,
could be utilized instead of WAX. However, this would affect the performance
and recovery of the ionic PFAS compounds.
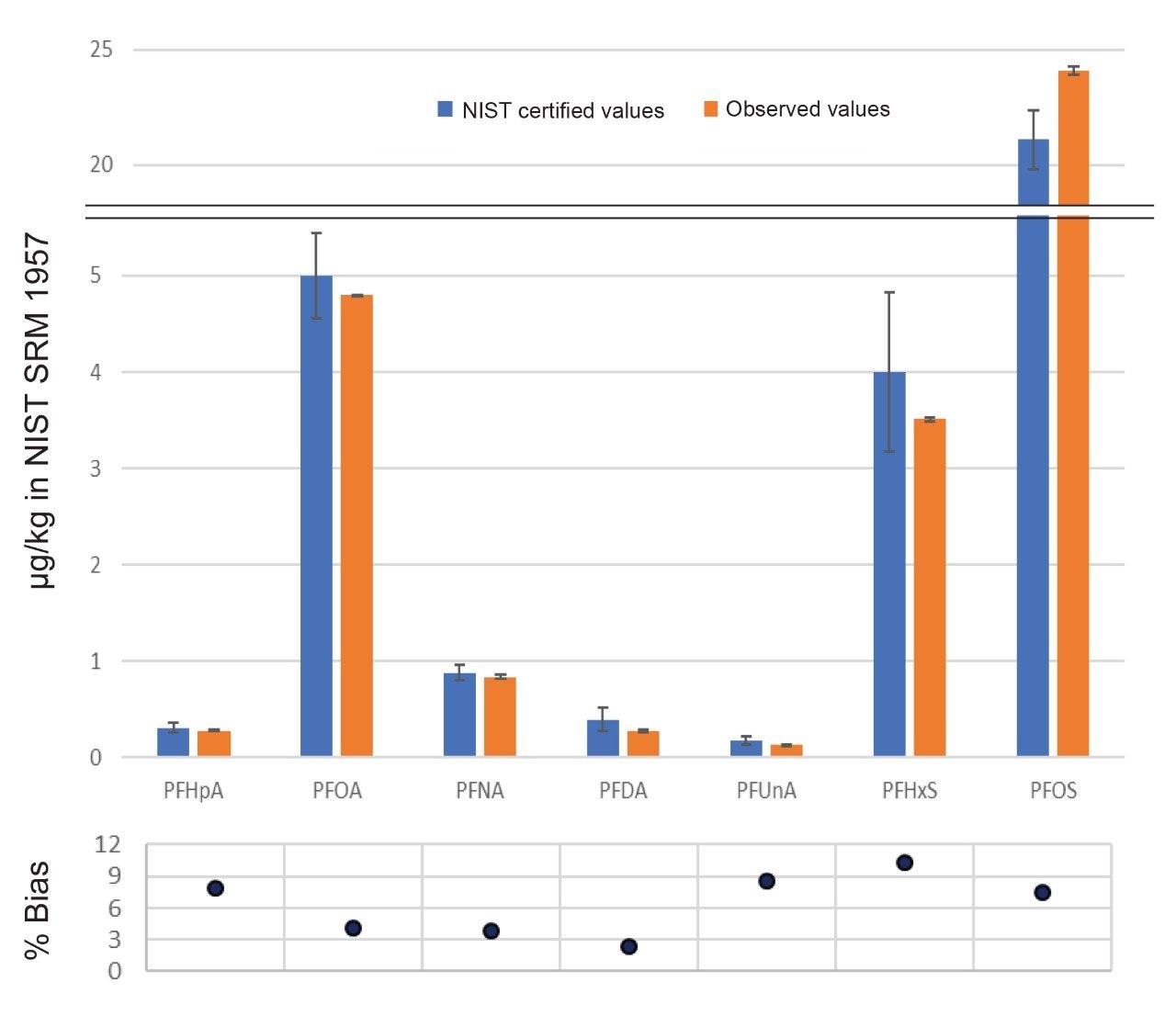
In addition to recovery, method performance was continually
evaluated through the use of a Standard Reference Material (SRM) from NIST.
Replicates of NIST SRM 1957, non-fortified human serum, were extracted with
every batch of samples to ensure consistent accurate results. This reference
material contains certified levels of seven PFAS naturally occurring in pooled
human serum, ranging in concentration from 0.172 to 21.1 µg/kg (or 0.043 to
5.27 ng/L). Since this SRM covers a concentration range of parts per trillion
(ppt) through parts per billion (ppb), it is a robust test of the methodology.
The accuracy and robustness of the method is demonstrated in Figure 6. The bar
graph shows the experimentally calculated concentration of eight replicates of
SRM 1957 extraction and analysis compared to the certified NIST values. The
scatter plot demonstrates the percent difference of the experimental results
from the certified range, with all measurements within ±10% of certified
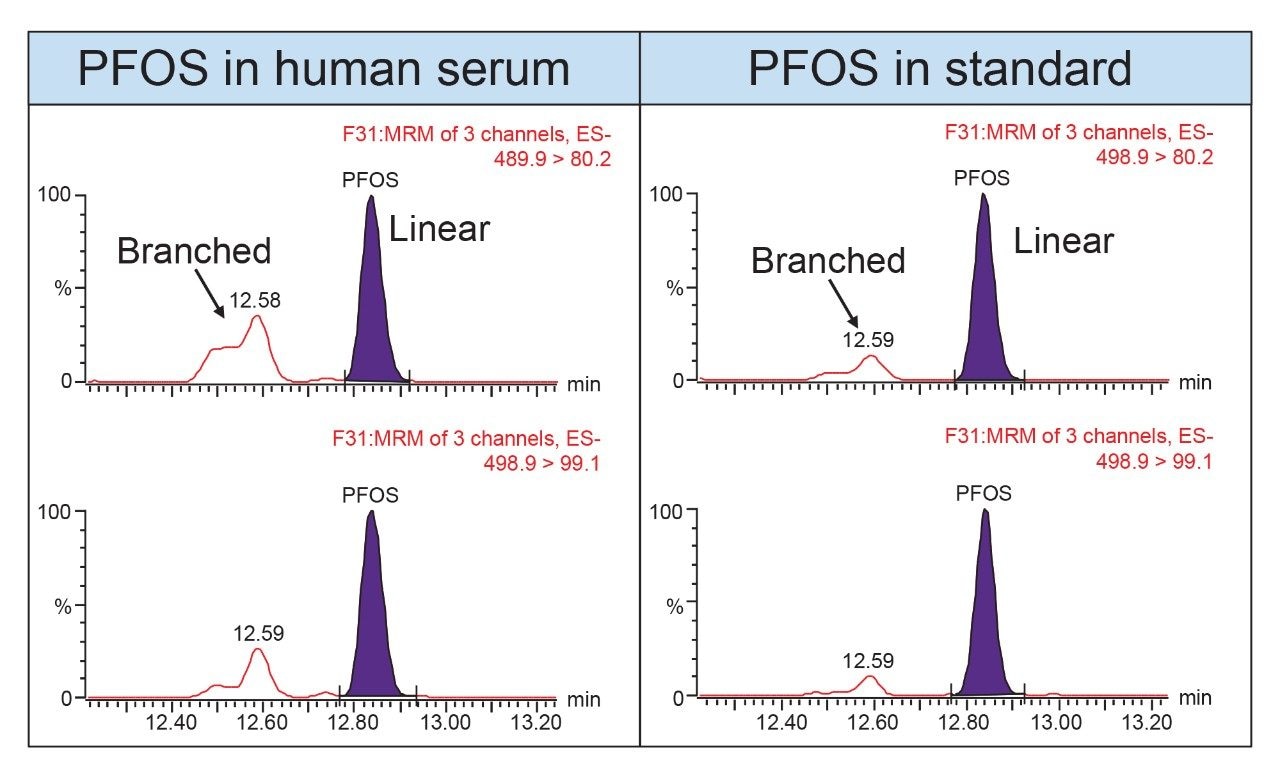
values. PFOS is the only compound that consistently produced slightly higher
experimental results, but this is most likely due to a difference in the way
the branched and linear isomers were handled during data processing.
Additionally, percent RSD of the eight replicates were all below 4%, indicating
the sample extraction and analysis is extremely robust.