Lipid Analysis Workflow Using a waters_connect™ DIA UPLC/MS Workflow With Xevo™ QTof G3
Nur zu Forschungszwecken. Nicht als diagnostisches Verfahren geeignet.
Abstract
The waters_connect platform features comprehensive software tools for the analysis and processing of LC-MS/MS based lipidomic data. Lipidomic workflows can incorporate multiple MS acquisition types such as data-dependent (DDA) and data-independent (DIA) acquisition datasets. Here we provide an evaluation of DIA UPLC/MS in combination with workflow-driven informatics for lipidomics research. Our findings demonstrate the significant capability of the waters_connect platform, which leverages Ultra Performance LC™ (UPLC™) technology coupled with the Xevo G3 QTof Mass Spectrometer for sample acquisition, identification, quantification, and reporting. Reproducible, accurate, and reliable identification of lipids was demonstrated by examination of corresponding chromatographic peaks and spectra. We established excellent linearity (R2 >0.97) for bioactive phospholipid calibration curves based on spiked standards in plasma, covering typical biological ranges. These calibration curves can be used to quantify endogenous lipids accurately which can then be reported in PDF or spreadsheet format. The data generated using the platform can also be transferred to third-party informatics using the unique application program interface (API) for processing and interpretation.
Benefits
- Simple and robust acquisition strategy, providing high-quality, comprehensive data quickly and efficiently
- Customizable workflows, allowing for ease of use and adaptation to different needs
- Highly flexible lipid identification approach, incorporating spectral matching, and in-source fragment ion recognition
- Easy and accurate quantitation using calibration curve capabilities
- Smart reporting of results using report templates in PDF or spreadsheets for transfer to statistical packages
- Compatibility with third-party tools such as Lipostar, MzMine, and Skyline, provides users with greater flexibility for data analysis
Introduction
Lipidomics, a maturing field of omics science, enables researchers to explore alterations in the lipidome that arise from disease, treatment, environmental exposure, and lifestyle, among other factors. The lipidome is complex, consisting of thousands of lipids spanning a broad concentration range, from polar free fatty acids and moderately polar bioactive lipids like lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), sphingomyelins, and ceramides, to nonpolar glycerol lipids such as triglycerides and cholesterol esters. Analyzing these lipids presents a daunting challenge, which typically requires a combination of reversed-phase liquid chromatography (RPLC) and precise mass spectrometry to perform open profiling experiments. Despite recent advancements in analytical technology, the detection, identification, and quantification of lipids of interest can be challenging due to data processing, database searching, and lipid identification.
Here we demonstrate the significant advantages of utilizing a complete system solution, consisting of an ACQUITY™ Premier UPLC, Xevo G3 QTof mass spectrometer and waters_connect informatics. The waters_connect software is a comprehensive software tool for the analysis and processing of LC-MS lipidomic data, which can incorporate multiple MS acquisition types such as data dependent (DDA) and data independent (DIA) acquisition datasets. Using waters_connect applications, samples can be submitted for analysis, processed and reported in the same workflow offering a streamlined, robust, reproducible and accurate means of identification, quantification, and reporting (Figure 1).
 Figure 1. Overview of the integrated waters_connect workflow from system setup through to data reporting.
Figure 1. Overview of the integrated waters_connect workflow from system setup through to data reporting.
Experimental
Avanti Odd-Chained LIPIDOMIX™ (Avanti, Birmingham, Al, USA) was spiked into NIST SRM 1950 plasma (Sigma Aldrich, Poole, UK) at three concentrations (10×, 20×, and 50×) using IPA. The neat standard mixture or spiking solutions were added at less than 5% v/v to commercially available pooled “normal plasma” (Peary Court, Novi, MI, USA) to generate a 10-point calibration curve.
Six replicates of NIST SRM 1950 plasma were tested. Both calibrants and test samples were prepared using the protein precipitation method described by Sarafian et al., 2014, where aliquots of plasma (25 μL) were transferred to low protein binding Eppendorf™ tubes followed by 125 μL of a 500-fold dilution of deuterated ceramide LIPIDOMIX™ (Avanti, Birmingham, Al, USA) and SPLASH LIPIDOMIX™ (Avanti, Birmingham, Al, USA) as internal standards in IPA/ACN (1:2, v/v) for protein precipitation.1
After vortex mixing and incubation at −20 °C for ten minutes, samples were shaken at 500 rpm on a Thermo-Shaker PCMT at 5 °C for two hours. The extracted samples were then centrifuged before transferring the supernatant to total recovery glass vials (Waters, Milford, MA, USA) for LC-MS/MS analysis.
LC Conditions
|
LC system: |
ACQUITY™ Premier Flow Through Needle (FTN) UltraPerformance LC |
|
Vials: |
Certified Glass Screw Neck Max Recovery Vials (p/n: 186000326c) |
|
Column(s): |
ACQUITY Premier UPLC CSH™ C18 2.1 x100 mm, 1.7 μm (p/n 186009461) |
|
Column temperature: |
55 °C |
|
Sample temperature: |
8 °C |
|
Injection volume: |
2 µL (ESI+); 2 µL (ESI-) |
|
Flow rate: |
0.4 mL/min |
|
Mobile phase A: |
600:390:10 Acetonitrile:Water:1M Ammonium Formate, 0.1% Formic Acid |
|
Mobile phase B: |
900:90:10 IPA:Acetonitrile:1M Ammonium Formate, 0.1% Formic Acid |
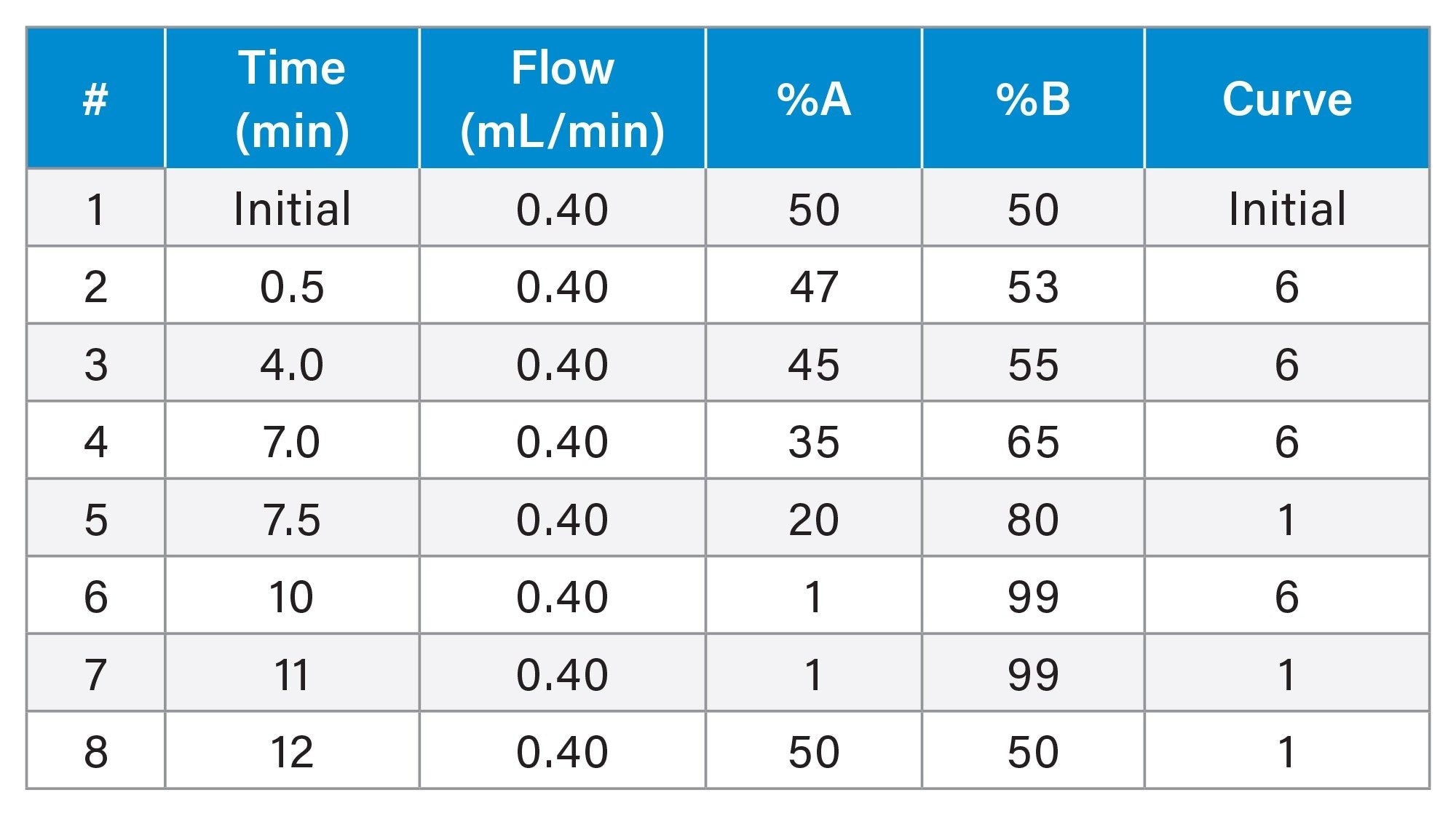
UPLC Gradient
MS Source Settings
|
Capillary voltage (kV): |
2.8 (+)/1.9 (-) |
|
Sampling cone (V): |
40 |
|
Source offset: |
30 |
|
Source temperature (°C): |
120 |
|
desolvation temperature (°C): |
500 |
|
cone gas flow (L/hr): |
150 |
|
Desolvation flow (L/hr): |
750 |
|
Detector auto gain: |
On |
|
Quad profile: |
Auto |
|
Lockspray flow (µL/min): |
20 |
Tof settings
|
MS function: |
Tof MSE |
|
Analyser mode: |
Resolution |
|
Dynamic range: |
Extended |
|
Mass range: |
50–1200 Da |
|
Scan time: |
0.1 secs |
|
Data format: |
Continuum |
|
Collision energy: |
20–45 eV |
|
Optional: |
Automatic Detector check |
|
Lockspray settings: |
Acquire lockspray & apply correction. Leucine Enkephalin acquired every 30 secs (scan time = 0.1 secs). |
Data were collected and processed using the UNIFI™ application in waters_connect. The Waters Lipidomics Profiling CCS Library was used to create the scientific libraries required for processing and compound identification in waters_connect.
Results and Discussion
System Setup and Acquisition
Automated detector set-up and instrument calibration are performed prior to the analysis of samples using the UNIFI application within waters_connect. This ensures the mass spectrometer is in good working order maximizing mass accuracy and response stability. Sets of samples can be intuitively generated using the UNIFI application embedded within waters_connect. Sample lists of calibrants, quality control, and test samples can be submitted with the required LC and MS conditions.
System Suitability
To ensure the integrity of the system, blank samples of IPA were injected to check for impurities and potential contamination. A neat standard mixture, diluted 100 times with EquiSPLASH in IPA, was used to verify the reproducibility of retention time and peak shape. As depicted in Figure 2, the resulting chromatograms of the deuterated lipid standards in positive and negative ESI modes provide an overview of the system's performance for lipidomic analysis.
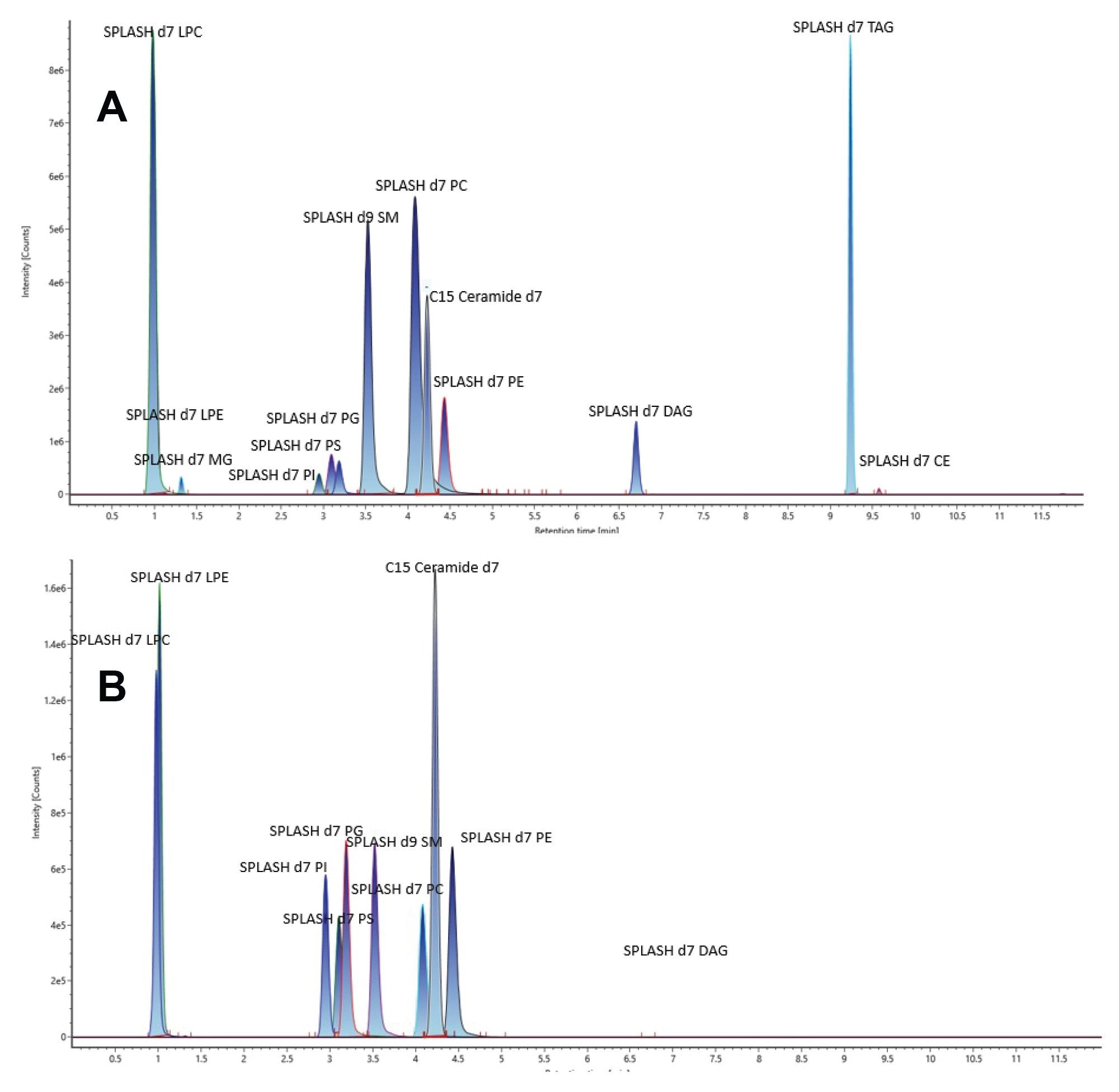 Figure 2. Chromatograms representing the Avanti EquiSPLASH; (A) Positive mode ESI (2 µL injection) and (B) Negative mode ESI (2 µL injection).
Figure 2. Chromatograms representing the Avanti EquiSPLASH; (A) Positive mode ESI (2 µL injection) and (B) Negative mode ESI (2 µL injection).
The retention times between both polarity modes were shown to be comparable and reproducible. Peak widths of 6.5 seconds and an overall peak capacity of 90 for a 10-minute separation were routinely achieved. These parameters have previously been shown to demonstrate suitable performance for lipidomic analysis of complex matrices.2
Identification
Lipid species to be identified or screened can be added to an “Analysis Method” from the scientific library such as the Waters Lipidomic Profiling CCS Library. The entire library can be used to add or select components for screening against specific lipids. Figure 3 shows representative spectra for an example deuterated PE lipid in positive and negative ESI mode. Low and high collision energy spectra are provided indicating the precursor and associated fragment ions respectively. The combination of mass accuracy, matched fragment ions, and isotopic fit all contribute to providing high confidence in the identifications returned.
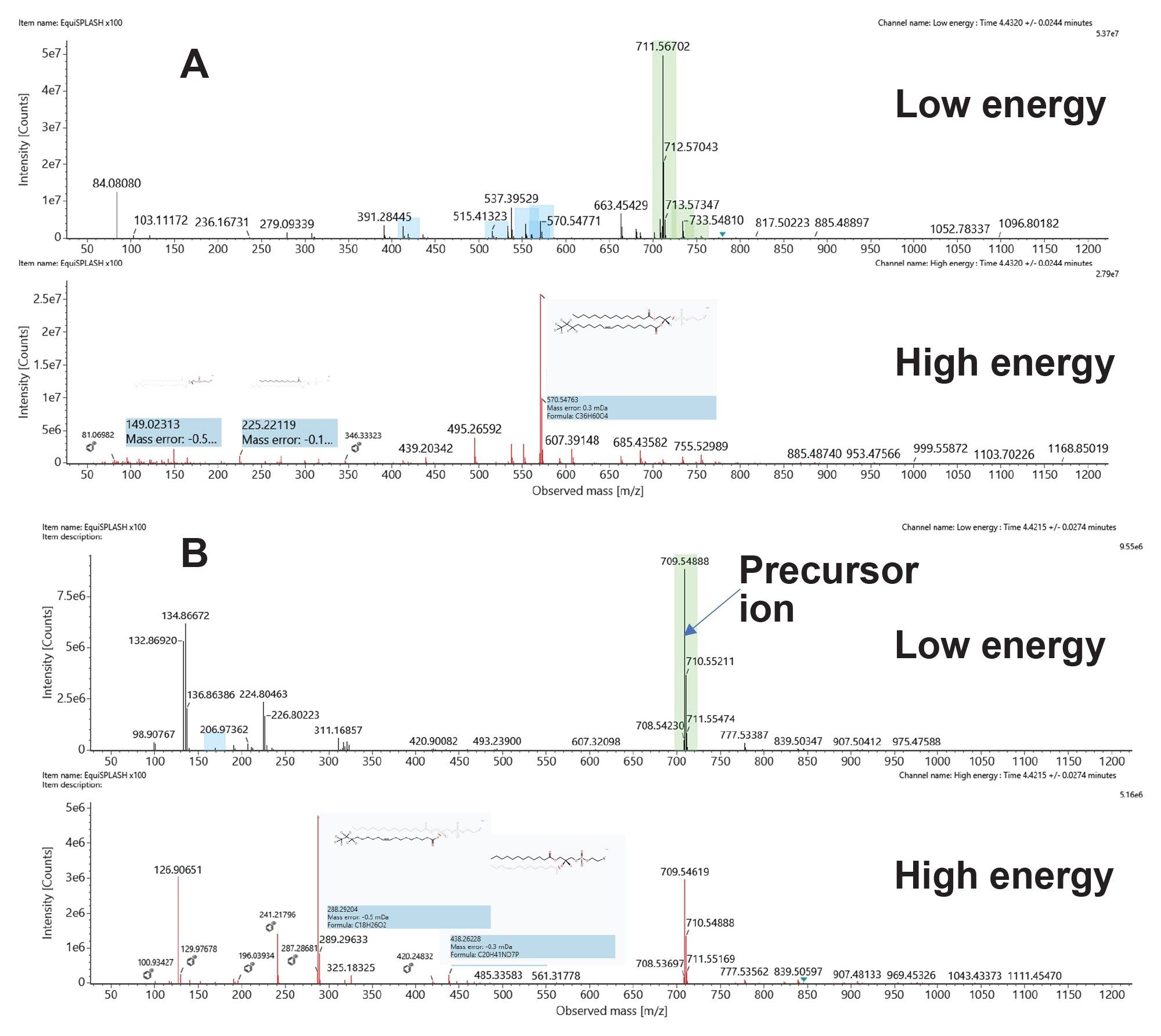 Figure 3. Low and high collision energy spectra of PE (d7); (A) Positive mode ESI and (B) Negative mode ESI.
Figure 3. Low and high collision energy spectra of PE (d7); (A) Positive mode ESI and (B) Negative mode ESI.
In Figure 4, we present both positive and negative ion chromatograms of an endogenous plasma lipid, identified as PC(16:0_18:2) with an elution time of 3.79 mins. In Figure 5A, we show the positive ion spectrum, which has a precursor mass error of +2.53 ppm. The high energy spectrum displays the typical PC head group of 184 m/z and various other theoretical fragments, suggesting a sum composition of PC(34:2). Complimentary negative ion mode (Figure 5B) data provides a high energy spectrum with more diagnostic fragment ions relating to the fatty acyl chains. The sn1 RCOO- ion fragment of 255.235 m/z confirms the presence of a 16:0 fatty acyl chain, and the sn2 RCOO- ion fragment at 279.235 m/z indicates the presence of an 18:2 fatty acyl chain. We can therefore assign the identification of PC(16:0_18:2) with a high degree of confidence.
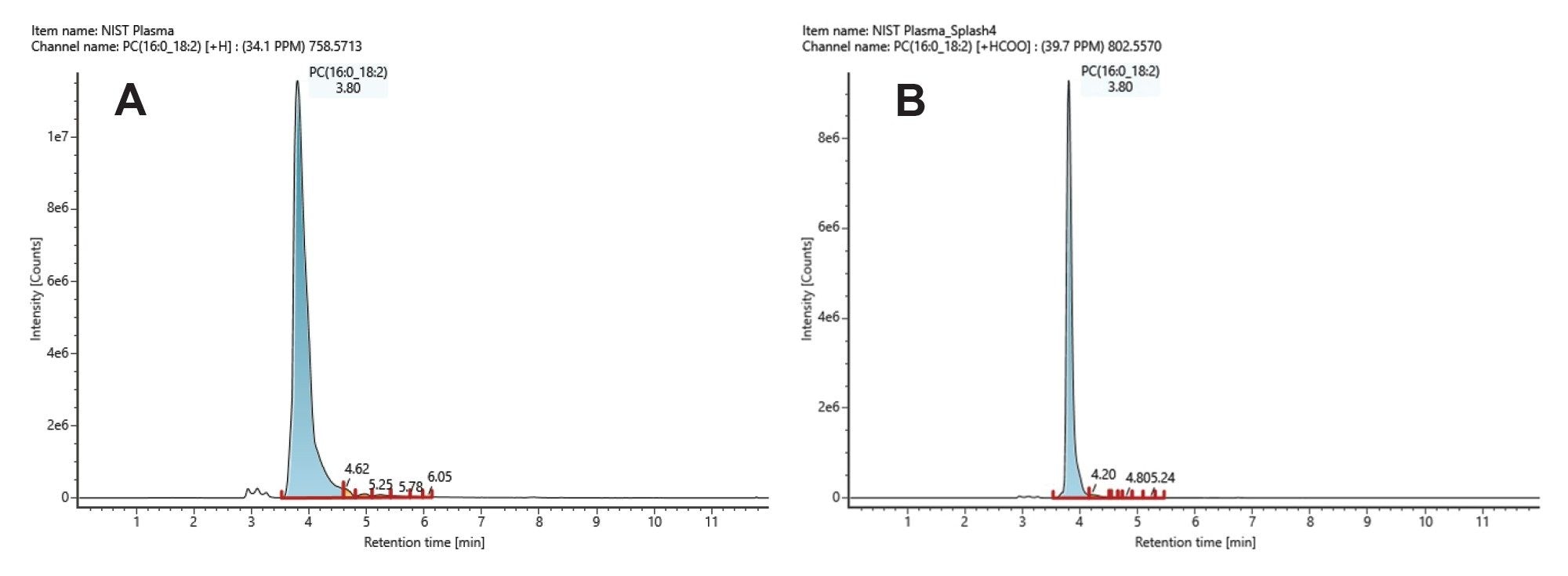 Figure 4. Positive (A) and negative (B) ion mode chromatograms of an endogenous plasma lipid, identified as PC(16:0_18:2).
Figure 4. Positive (A) and negative (B) ion mode chromatograms of an endogenous plasma lipid, identified as PC(16:0_18:2).
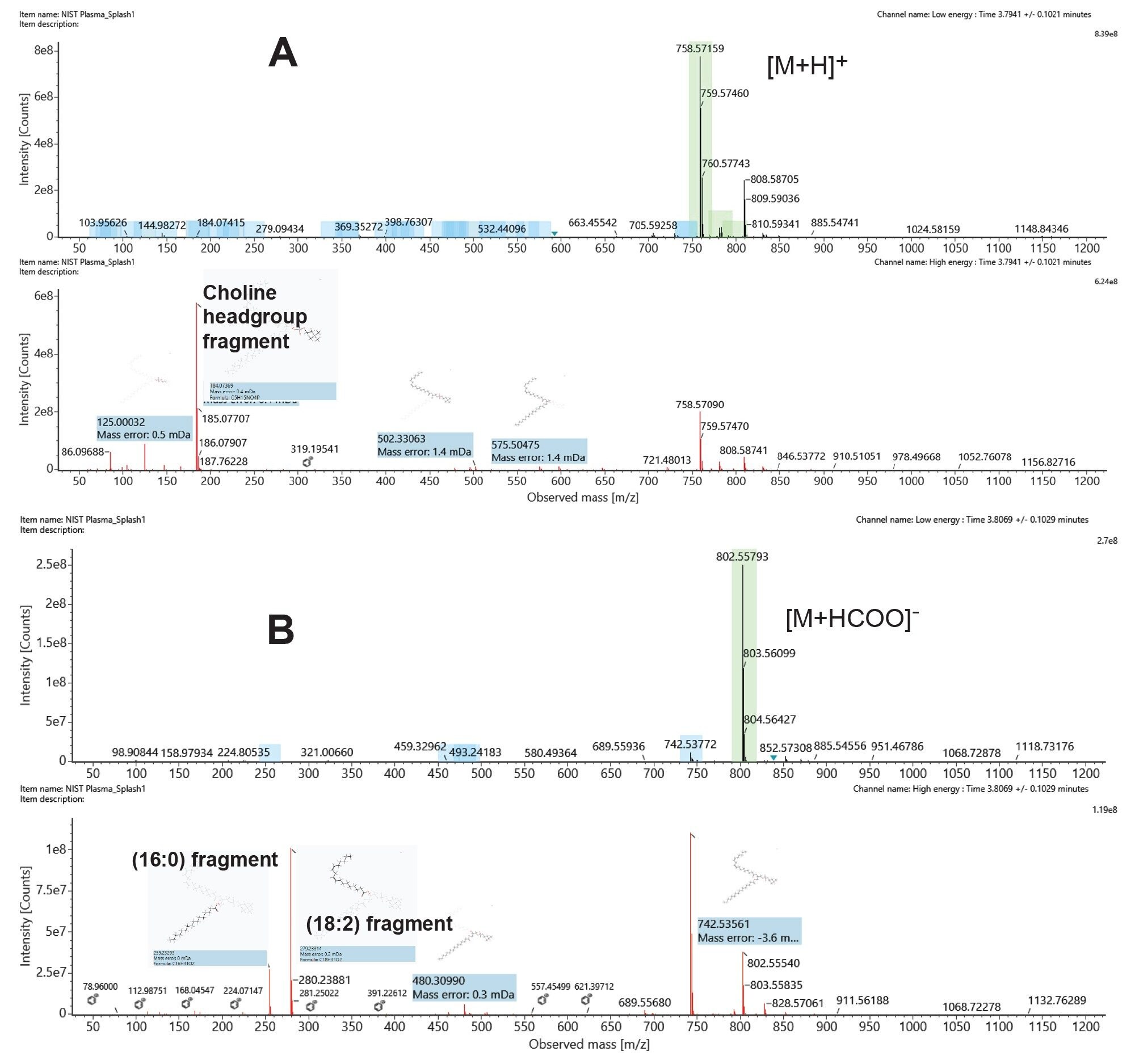 Figure 5. Low and high collision energy spectra of PC (16:0_18:2); (A) Positive mode ESI and (B) Negative mode ESI.
Figure 5. Low and high collision energy spectra of PC (16:0_18:2); (A) Positive mode ESI and (B) Negative mode ESI.
Quantification
Quantitative data is an essential component of lipidomic studies that enable accurate identification and quantification of lipid species. In LC-MS-based lipidomics, the ability to generate reliable quantitative data is dependent on the quality of the analytical method used. One way to evaluate the quantitative performance of an LC-MS method is to use calibration curves generated from standard lipid mixtures (i.e., odd chain mix) of known concentration. This standard mix covers the expected concentrations of lipid classes present in plasma or serum samples.3
The linearity of the calibration curves was evaluated using the R2 value, which indicates the degree of correlation between the analyte concentration and the signal intensity. Figure 6 shows example calibration curves of lipid standards in both positive and negative ion modes, spanning a dynamic range of three orders. The phospholipids, for example, showed good linearity (R2>0.97) for the concentration ranges tested (Table 1) and in agreement with previous tandem quadrupole, absolute quanitfication experiments.4
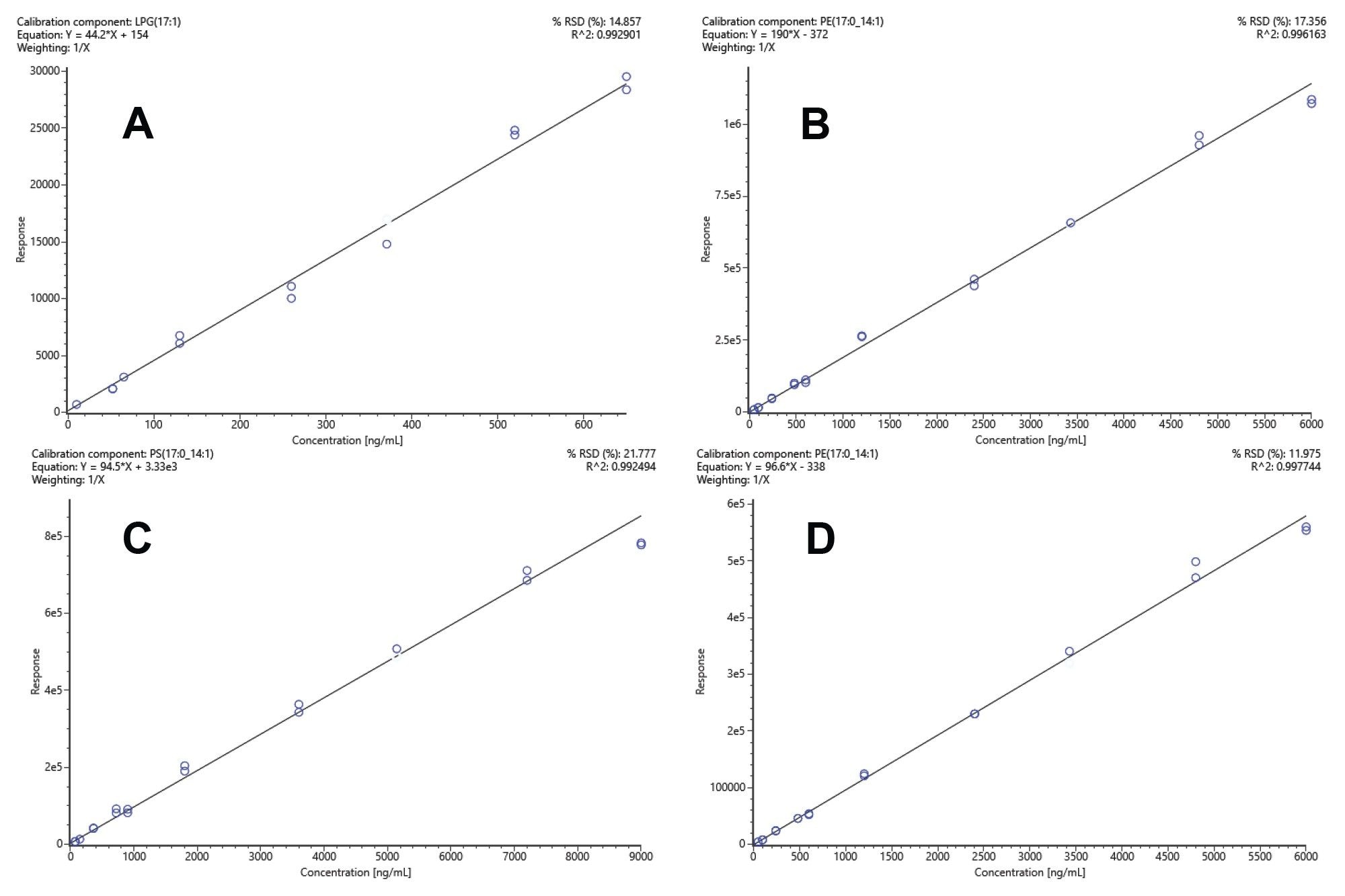 Figure 6. Example standard calibration curves representing LPG (ESI+) R2= 0.99 (A), PE (ESI+) R2= 0.99 (B), PS (ESI-) R2= 0.99 (C), PE (ESI-) R2= 0.99 (D). In all cases, a weighting of 1/x was applied.
Figure 6. Example standard calibration curves representing LPG (ESI+) R2= 0.99 (A), PE (ESI+) R2= 0.99 (B), PS (ESI-) R2= 0.99 (C), PE (ESI-) R2= 0.99 (D). In all cases, a weighting of 1/x was applied.
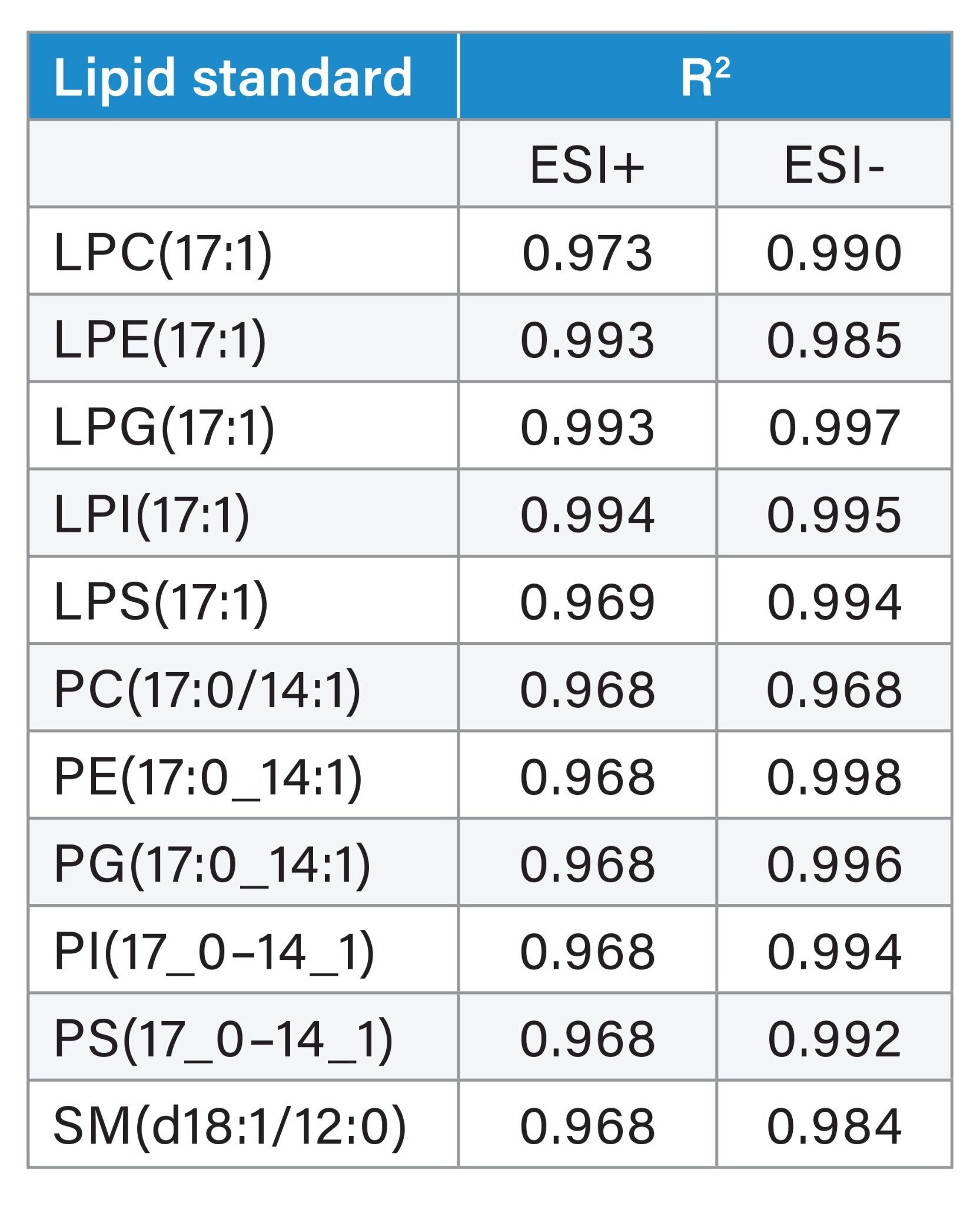 Table 1. Calibration curve R2 values generated using Odd Chain lipid mix spiked into plasma.
Table 1. Calibration curve R2 values generated using Odd Chain lipid mix spiked into plasma.
By examining the response of the lipids in test samples (i.e., NIST Plasma) and locating the corresponding point on the linear regression line (as shown in Figure 7), the concentration of the lipid species can be determined within UNIFI. For instance, the mean response for LPE(16:0) in the six test samples was approximately 75,000, which translates to a concentration of 300 ng/mL or 0.6 µM. This falls within the range of consensus concentration reported for this specific lipid species from previously reported studies.3 Table 2 shows the calculated concentration values of endogenous lipids using the calibration curves generated within UNIFI. The reported concentrations show good reproducibility, generating CVs <10%. The more abundant lipids such as LPC, PC, and SM show CV <3%.
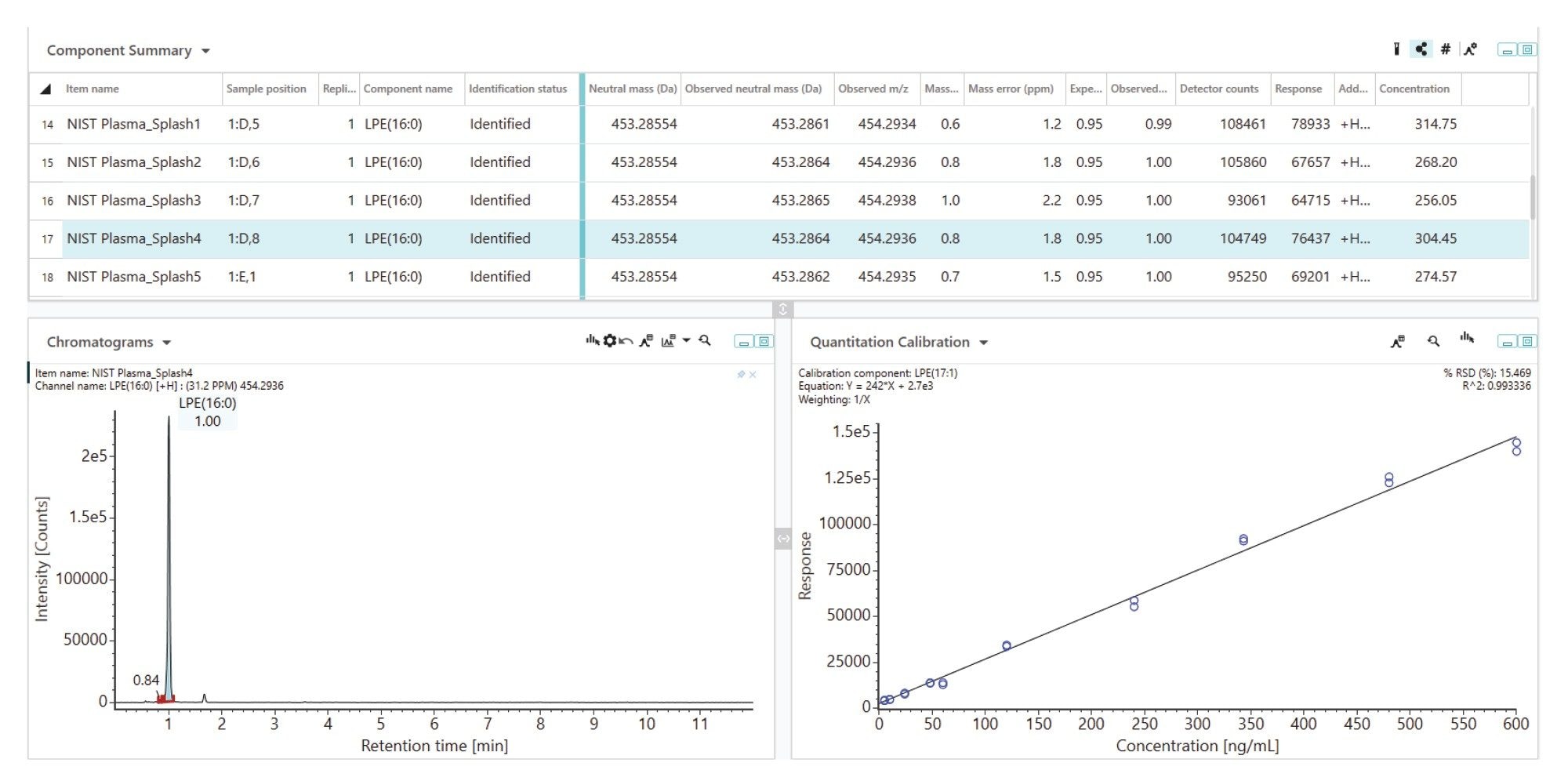 Figure 7. Example calibration curve for the spiked LPE standard (ESI+), which can be used to quantify the endogenous LPE by comparing the responses from the test samples (NIST SRM 1950 Plasma) to the linear regression line. The data table shows the mass accuracy, retention time, adducts, responses, and concentrations.
Figure 7. Example calibration curve for the spiked LPE standard (ESI+), which can be used to quantify the endogenous LPE by comparing the responses from the test samples (NIST SRM 1950 Plasma) to the linear regression line. The data table shows the mass accuracy, retention time, adducts, responses, and concentrations.
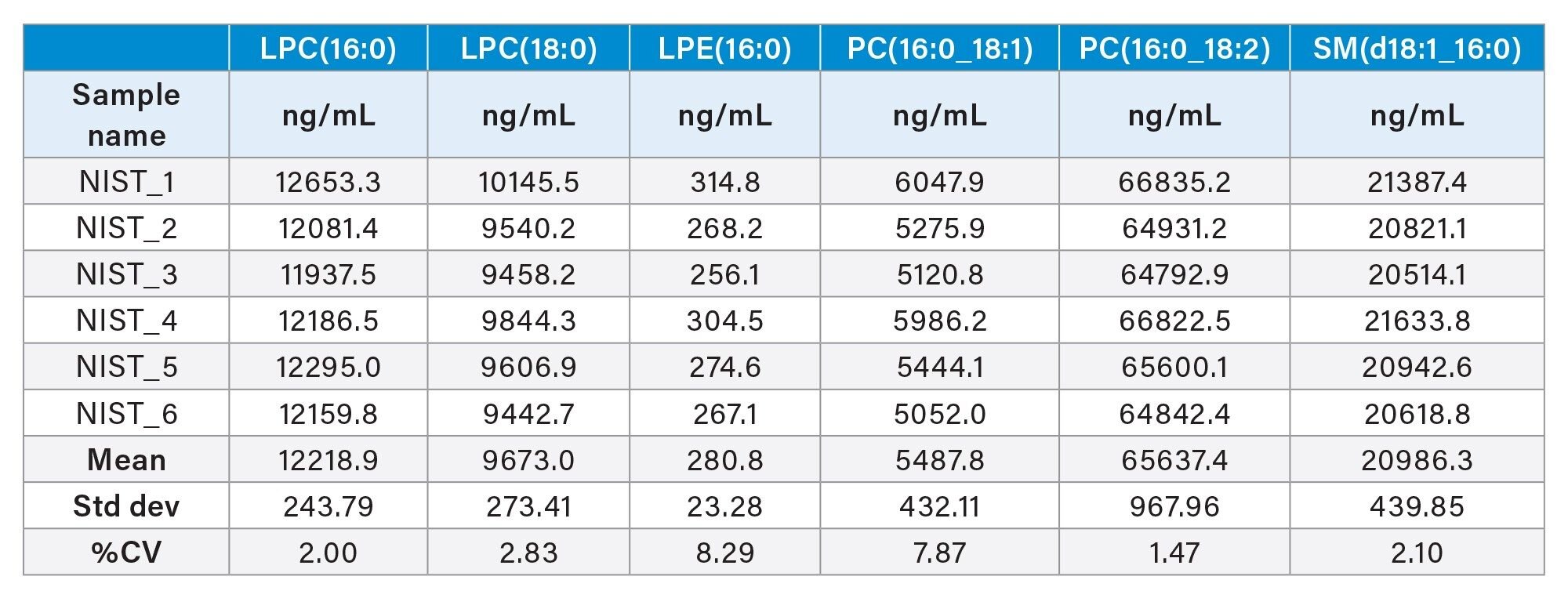 Table 2. Selection of calculated concentrations of endogenous plasma lipids quantified using calibration curves with the waters_connect workflow.
Table 2. Selection of calculated concentrations of endogenous plasma lipids quantified using calibration curves with the waters_connect workflow.
Overall, the results demonstrate the use of a QTof DIA methodology for generating quantitative lipidomic data. The quality of the data obtained can be improved by using validated calibration curves and optimising the sample preparation and analysis protocols. The use of internal standards and stable isotope-labelled standards can also help to improve the accuracy and precision of the quantification method.
Reporting
Results can be reported using the various templates available in UNIFI. These templates can be modified to suit the user’s needs. Figure 8, shows an example of a customized report and a default report for calculated concentrations. Reports can be generated as PDF or spreadsheet formats to enable multivariate analysis using third-party software such as MetaboAnalyst.5
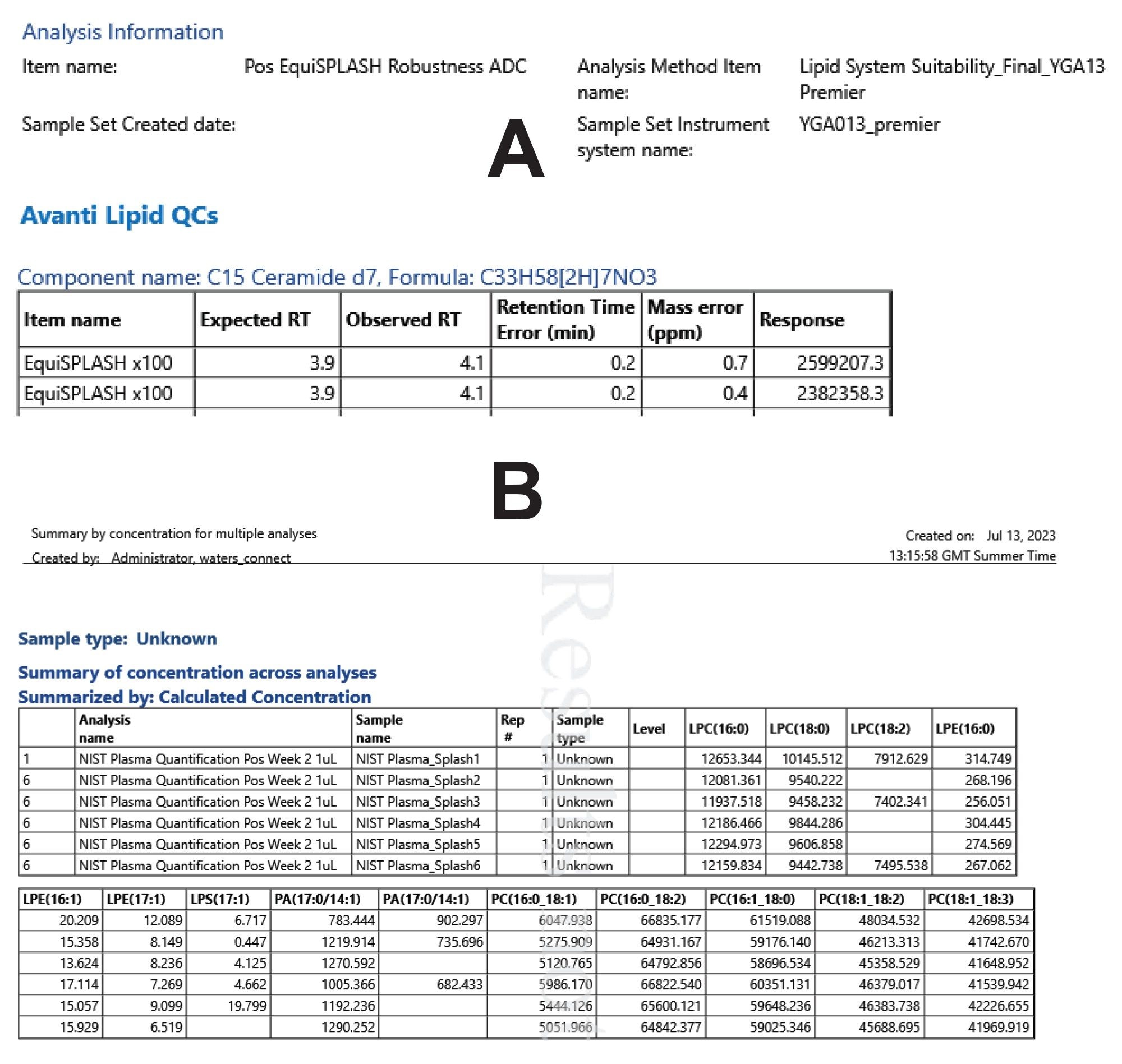 Figure 8. Example report templates for reporting data directly out of the UNIFI application can be customized (A) e.g. system suitability report or used as default (B) e.g. summary of concentrations across analyses.
Figure 8. Example report templates for reporting data directly out of the UNIFI application can be customized (A) e.g. system suitability report or used as default (B) e.g. summary of concentrations across analyses.
Furthermore, data generated within the UNIFI application can be accessed directly and processed with third-party software such as Lipostar using the unique application program interface (API) as shown in Figure 9.6
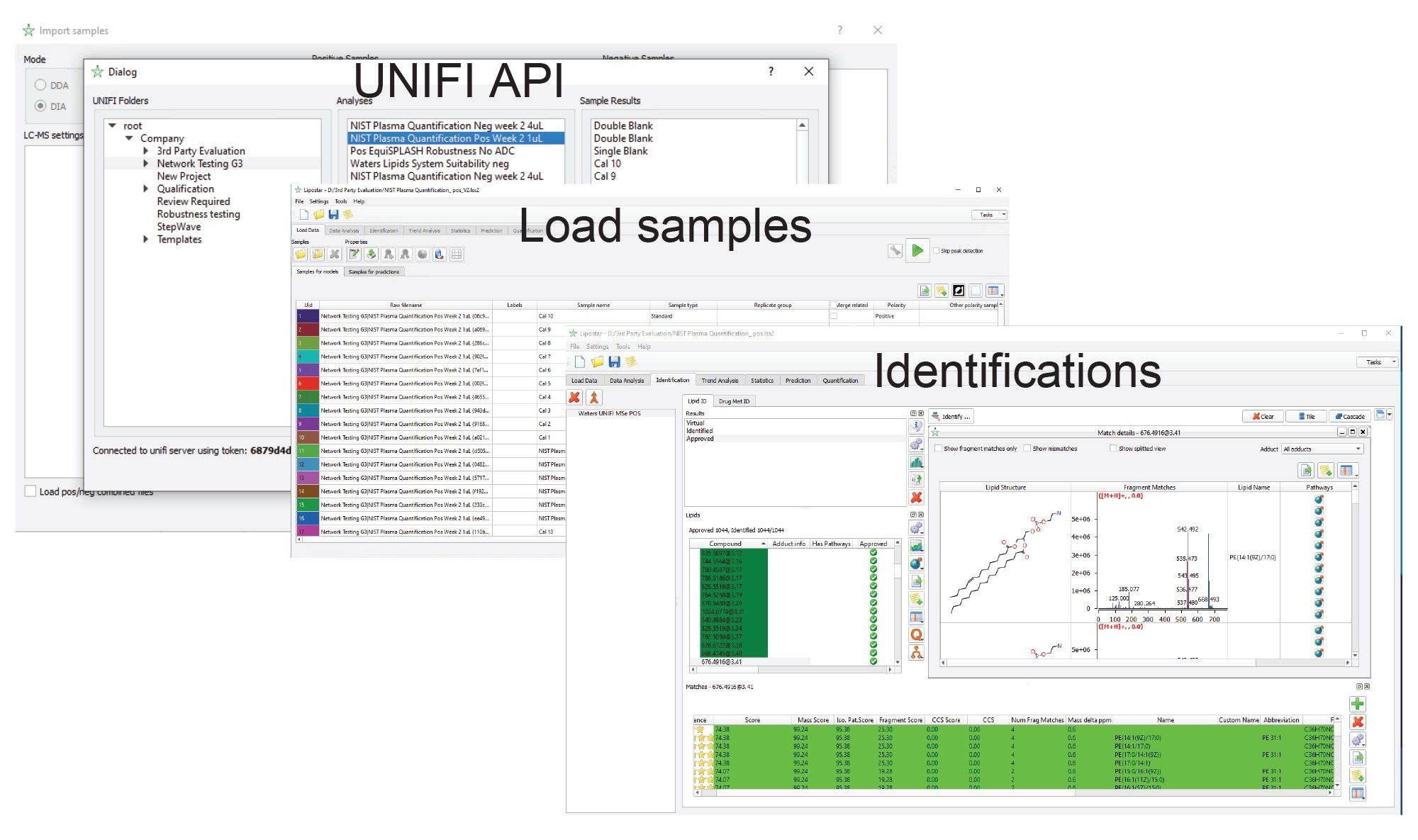 Figure 9. The application program interface can be used to transfer and process data to third-party software packages such as Lipostar.
Figure 9. The application program interface can be used to transfer and process data to third-party software packages such as Lipostar.
Conclusion
In summary, we have highlighted the exceptional features and advantages of a UPLC-DIA workflow for accurate mass profiling. We demonstrate how robust and high-quality lipidomics data can be easily generated, processed, interrogated and reported using the available workflows within waters_connect. Additional flexibility is enabled by third-party software compatibility.
References
- Sarafian, M. H.; Gaudin, M.; Lewis, M. R.; Martin, F.-P.; Holmes, E.; Nicholson, J. K.; Dumas, M.-E. Objective Set of Criteria for Optimization of Sample Preparation Procedures for Ultra-High Throughput Untargeted Blood Plasma Lipid Profiling by Ultra Performance Liquid Chromatography-Mass Spectrometry. Anal. Chem. 2014, 86, 5766–5774, DOI: 10.1021/ac500317c
- Giorgis Isaac, Nyasha Munjoma, Lee A. Gethings, Lauren Mullin, Robert S. Plumb. Waters Application note, 720006959, July 2020.
- Burla, B.; Arita, M.; Arita, M.; Bendt, A. K.; Cazenave-Gassiot, A.; Dennis, E. A.; Ekroos, K.; Han, X.; Ikeda, K.; Liebisch, G.; Lin, M. K.; Loh, T. P.; Meikle, P. J.; Orešič, M.; Quehenberger, O.; Shevchenko, A.; Torta, F.; Wakelam, M. J. O.; Wheelock, C. E.; Wenk, M. R. MS-based Lipidomics of Human Blood Plasma - a Community-initiated Position Paper to Develop Accepted Guidelines. J. Lipid Res. 2018, 59, 2001–2017, DOI: 10.1194/jlr.S087163
- Nyasha Munjoma, Giorgis Isaac, Ammara Muazzam, Olivier Cexus, Fowz Azhar, Hardev Pandha, Anthony D. Whetton, Paul A. Townsend, Ian D. Wilson, Lee A. Gethings, and Robert S. Plumb. High Throughput LC-MS Platform for Large Scale Screening of Bioactive Polar Lipids in Human Plasma and Serum, Journal of Proteome Research 2022 21 (11), 2596–2608, DOI: 10.1021/acs.jproteome.2c00297
- Chong, J., Soufan, O., Li, C., Caraus, I., Li, S., Bourque, G., Wishart, D.S., Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucl. Acids Res., 2018, 46, W486–494.
- Laura Goracci, Sara Tortorella, Paolo Tiberi, Roberto Maria Pellegrino, Alessandra Di Veroli, Aurora Valeri, and Gabriele Cruciani (2017) LipoStar, a Comprehensive Platform-Neutral Cheminformatics Tool for Lipidomics, Analytical Chemistry 2017 89 (11), 6257–6264, DOI: 10.1021/acs.analchem.7b01259
Featured Products
720008111, December 2023