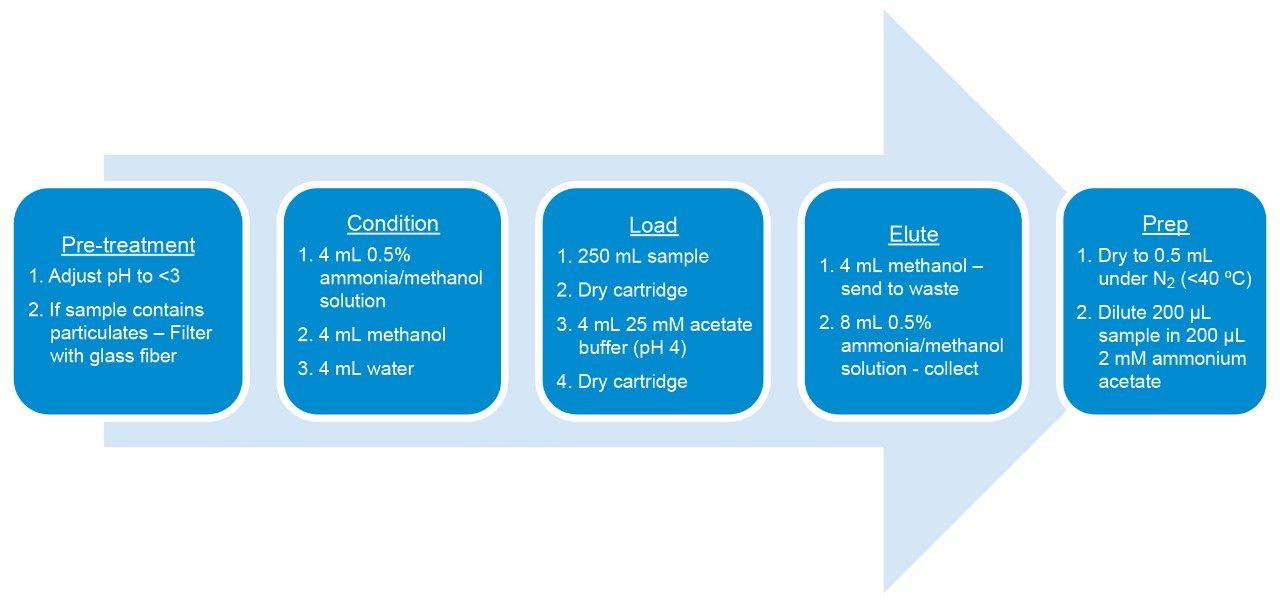
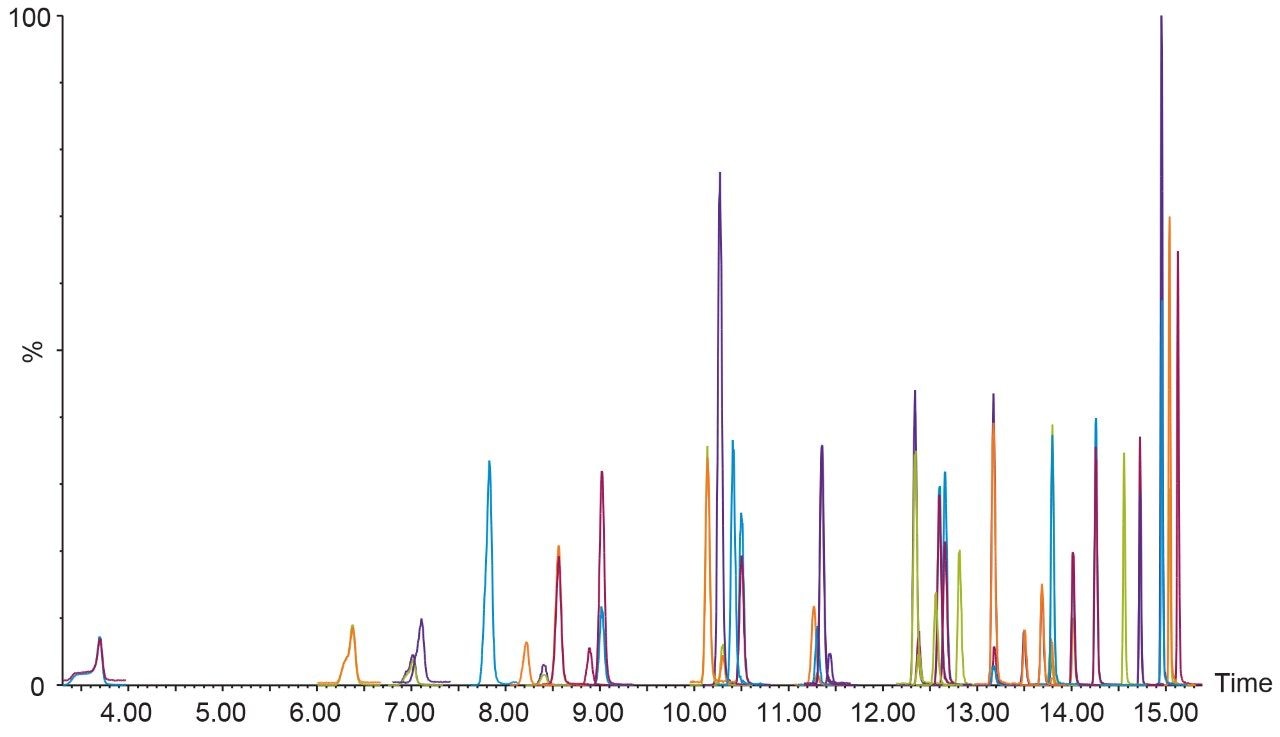
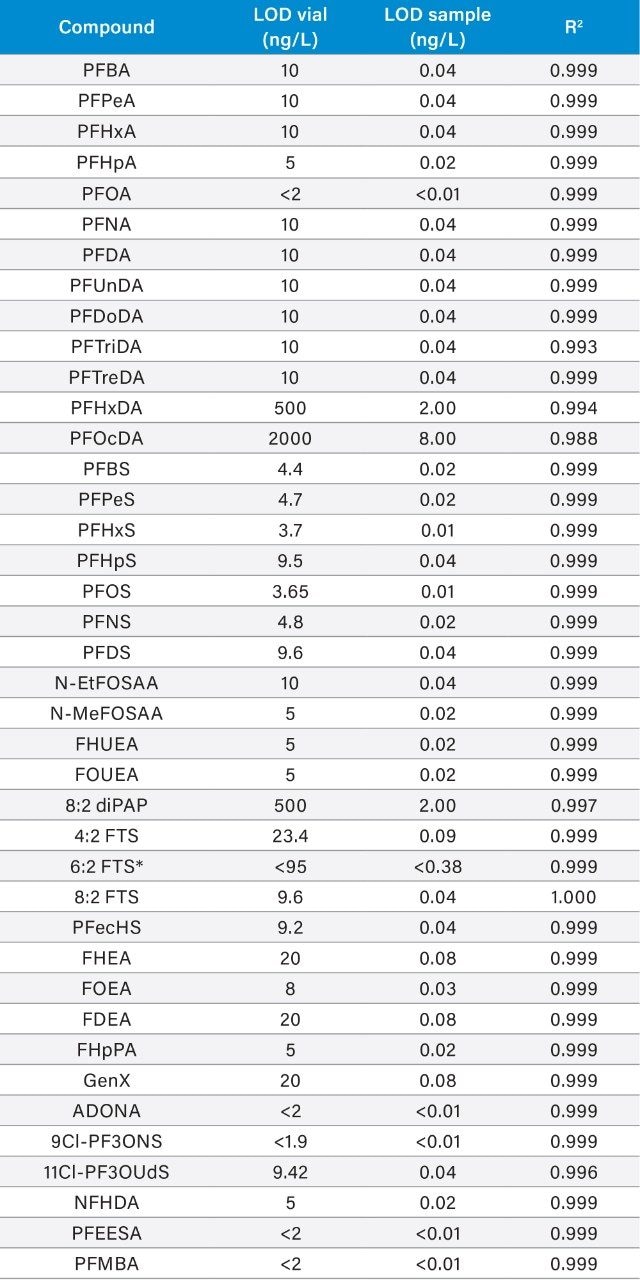
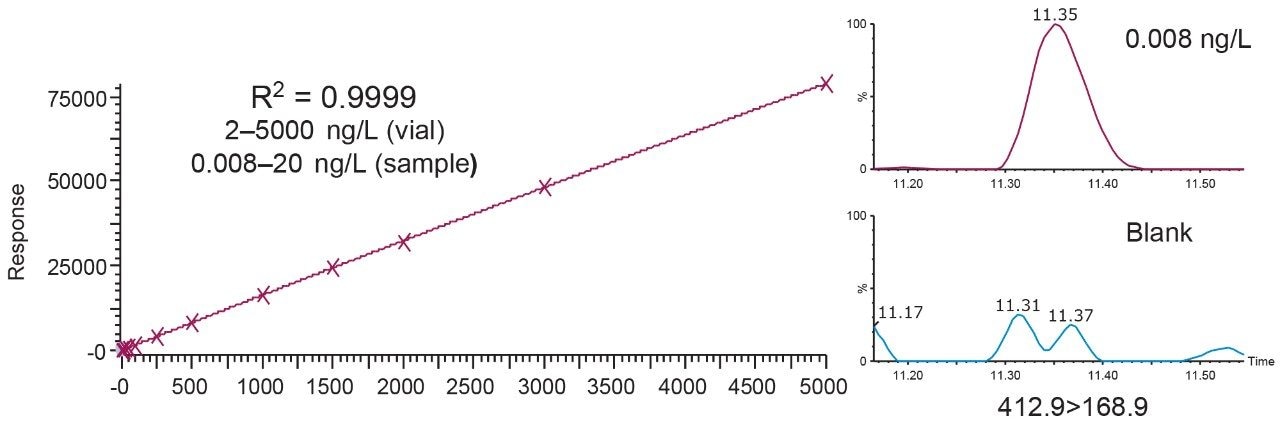
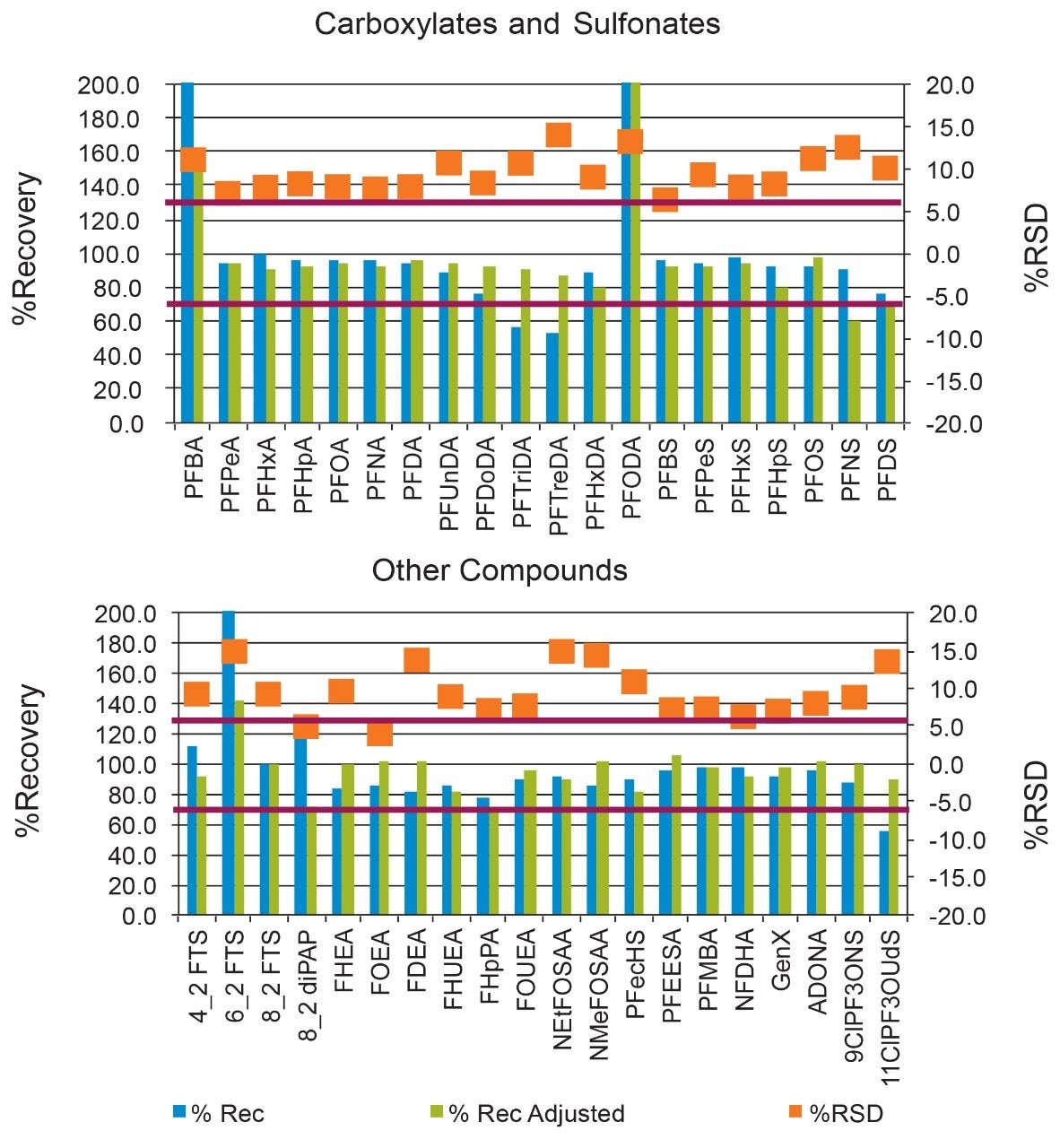
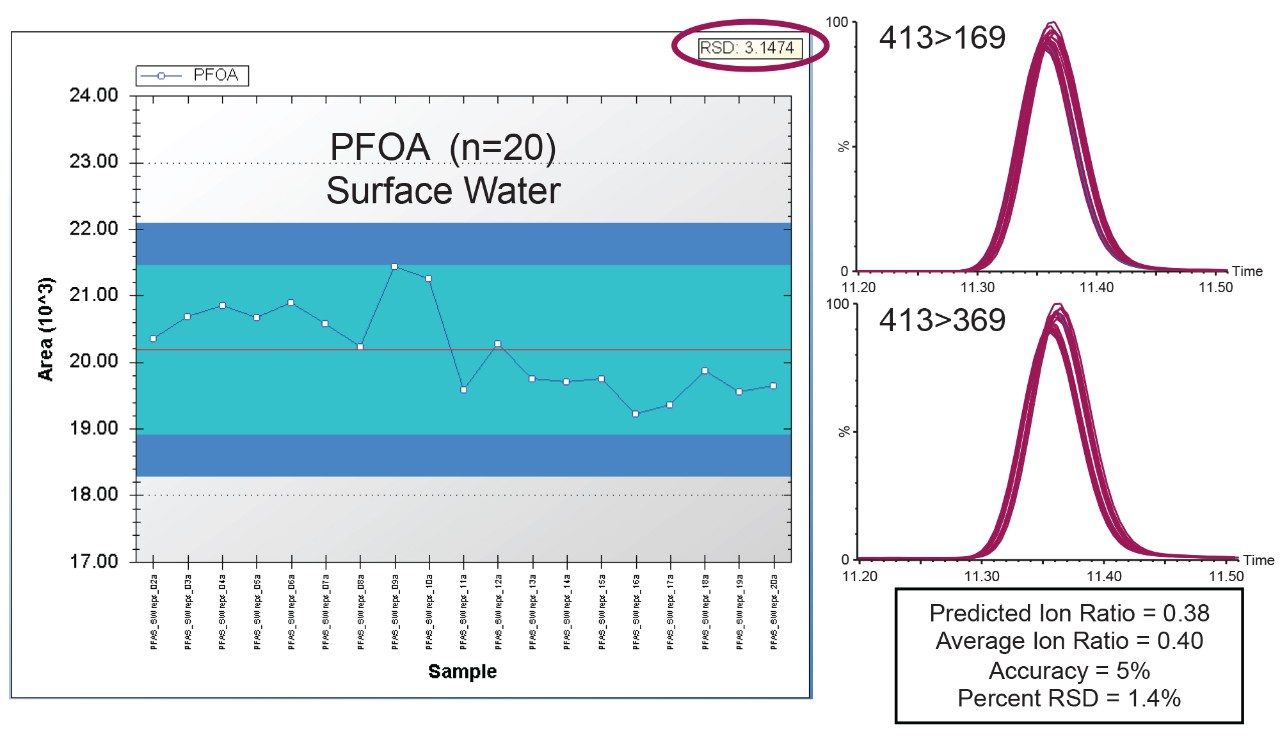
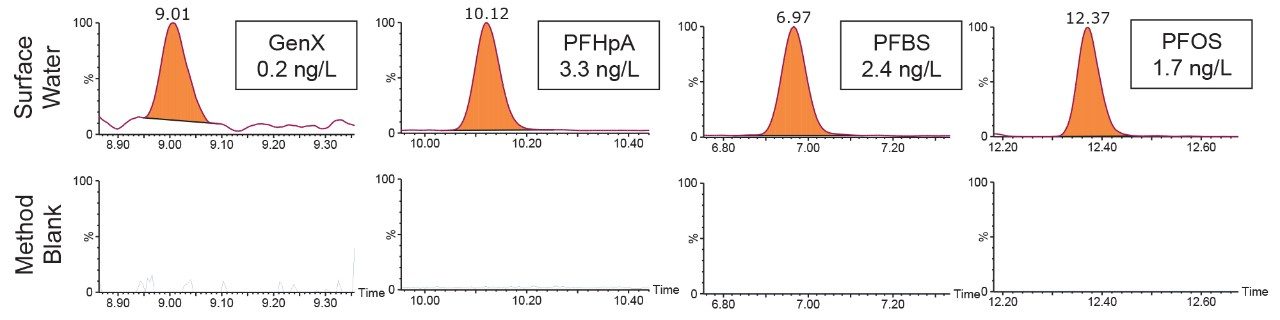
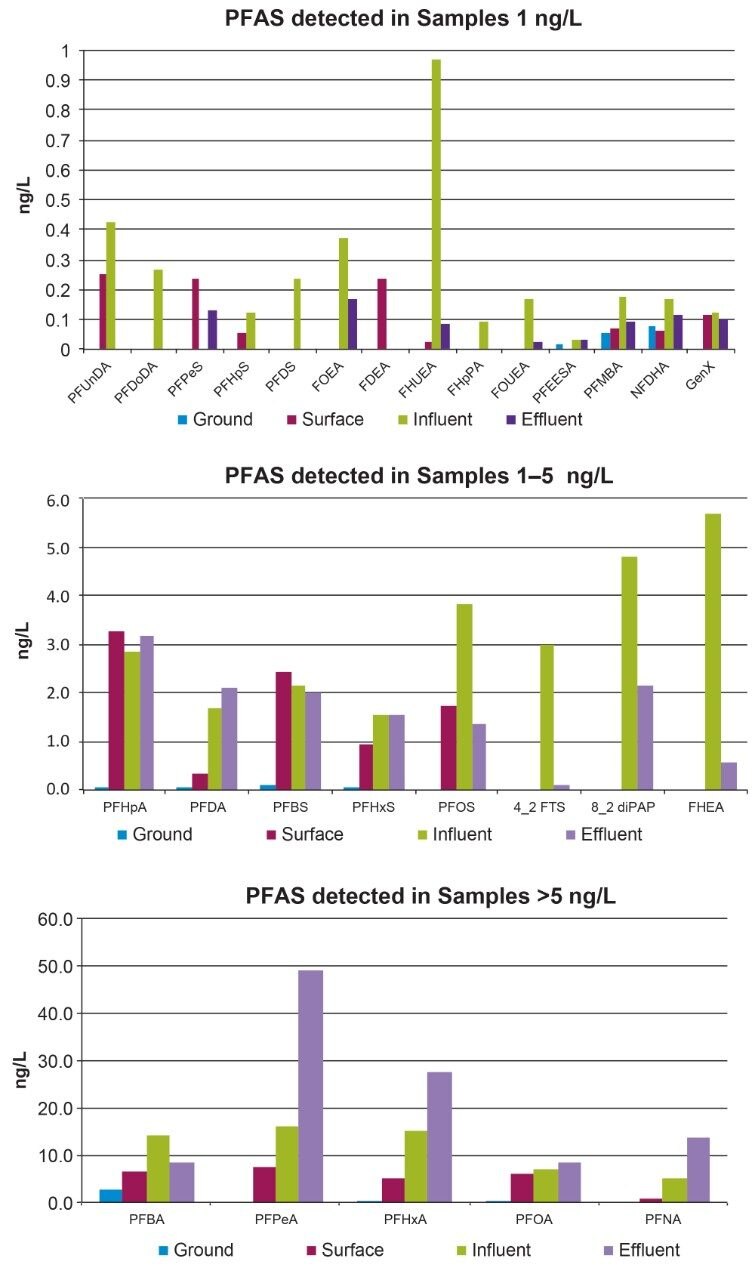
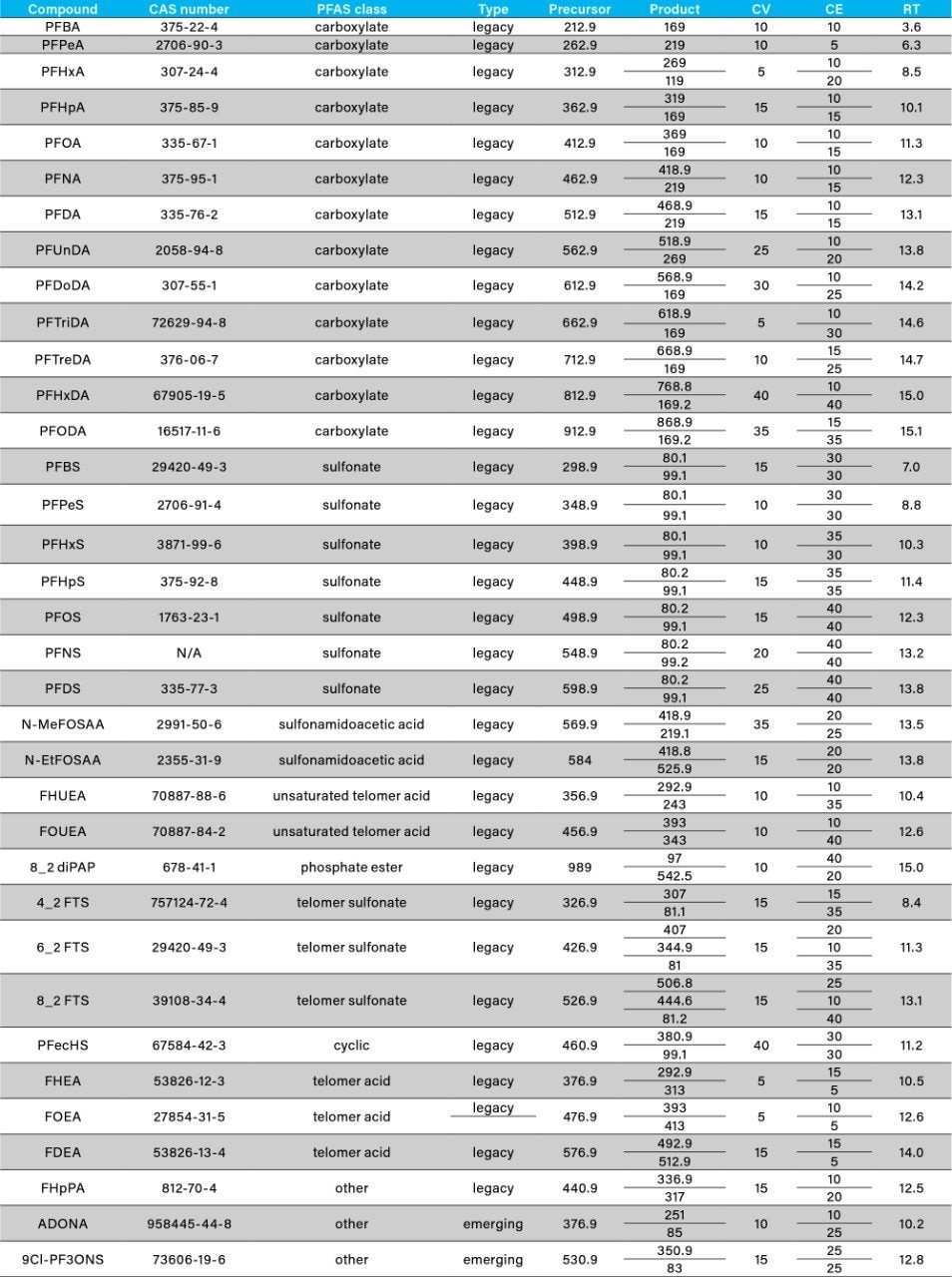
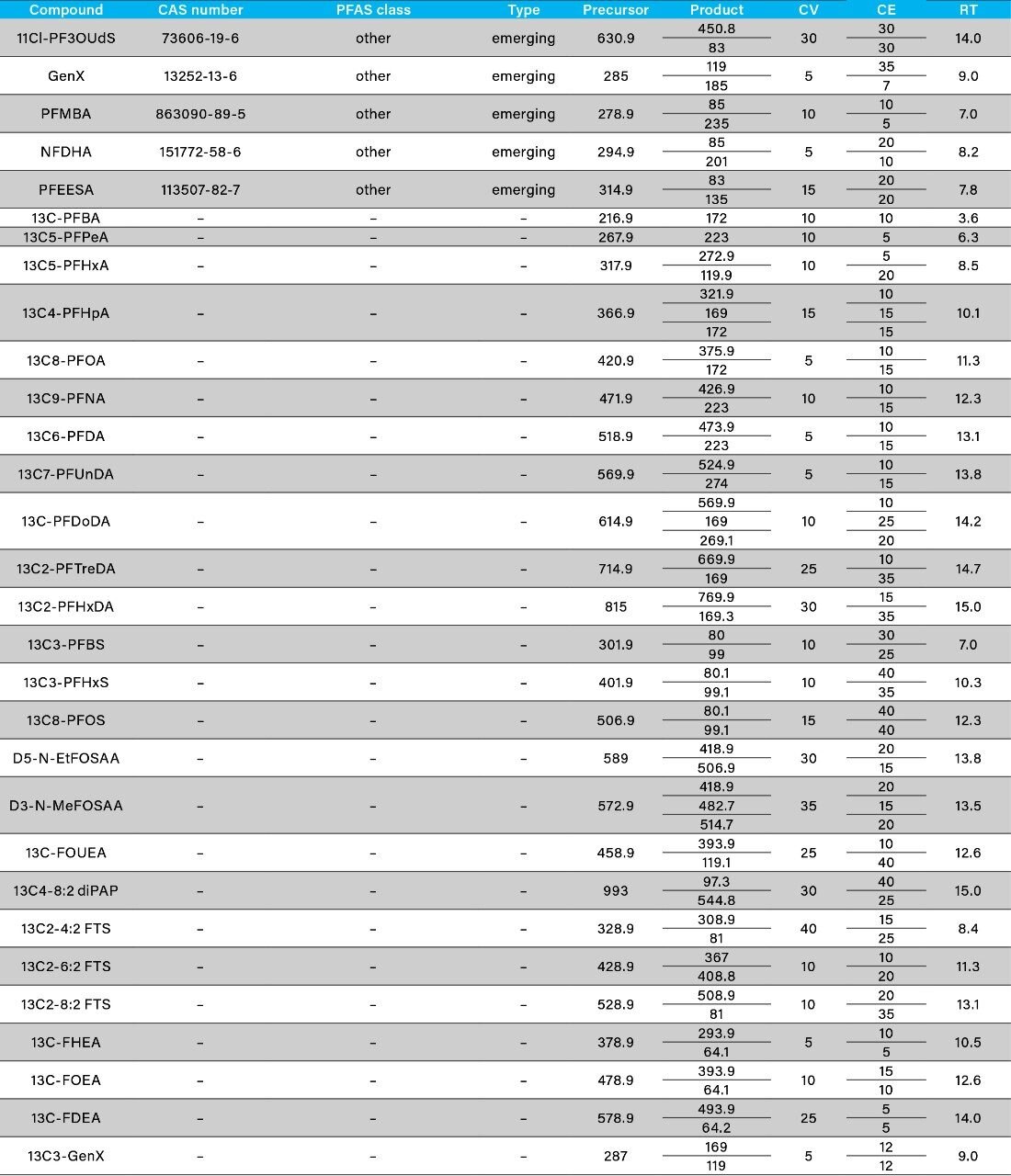
The ISO 25101 method was utilized as a guideline for the sample preparation methodology used for this analysis. Currently, ISO 25101 covers the extraction and analysis of only PFOA and PFOS. For this method, an extended list of PFAS compounds were considered and added. Appendix A contains information on all of the PFAS compounds analyzed in this method, together with a subset of emerging compounds being used to replace the legacy PFAS compounds, including GenX. All standards were obtained from Wellington Laboratories (Guelph, Ontario).
A Certified QC Standard (cat no.: 731) from ERA (Golden, CO), for use with ground and surface waters, was utilized as an instrumental QC check throughout the analysis. The standard contained a mix of 12 PFAS compounds. Certified values and QC Performance Acceptance Limits for each compound in the mix are provided with the standard, making instrumental QC evaluation quick and straightforward.
Due to widespread use of PFAS substances there are many common sources of potential contamination to the analysis. Since required detection limits are in the low- to sub-ng/L, care must be taken during sample collection, preparation, and analysis. Considering there are many common sources of PFAS contamination in the field and laboratory, it is recommended that any laboratory supplies to be used for this analysis be checked for PFAS contamination before use, as is practical. Contamination is also unavoidable from the chromatographic system. Therefore steps should be taken to minimize any system contribution, and as such, the Waters PFC Analysis Kit (p/n: 176001744) for the UPLC system was utilized. The kit is comprised of PFAS-free components (such as PEEK tubing to replace the conventional Teflon coated solvent lines) and an isolator column that helps to delay any residual background interferences from co-eluting with the analytical peak. Installation of the PFC Analysis Kit is straightforward and quick.5 In addition, special mobile phase solvents from Honeywell (Muskegon, MI) were used that were bottled in a manner to reduce residual background PFAS levels.