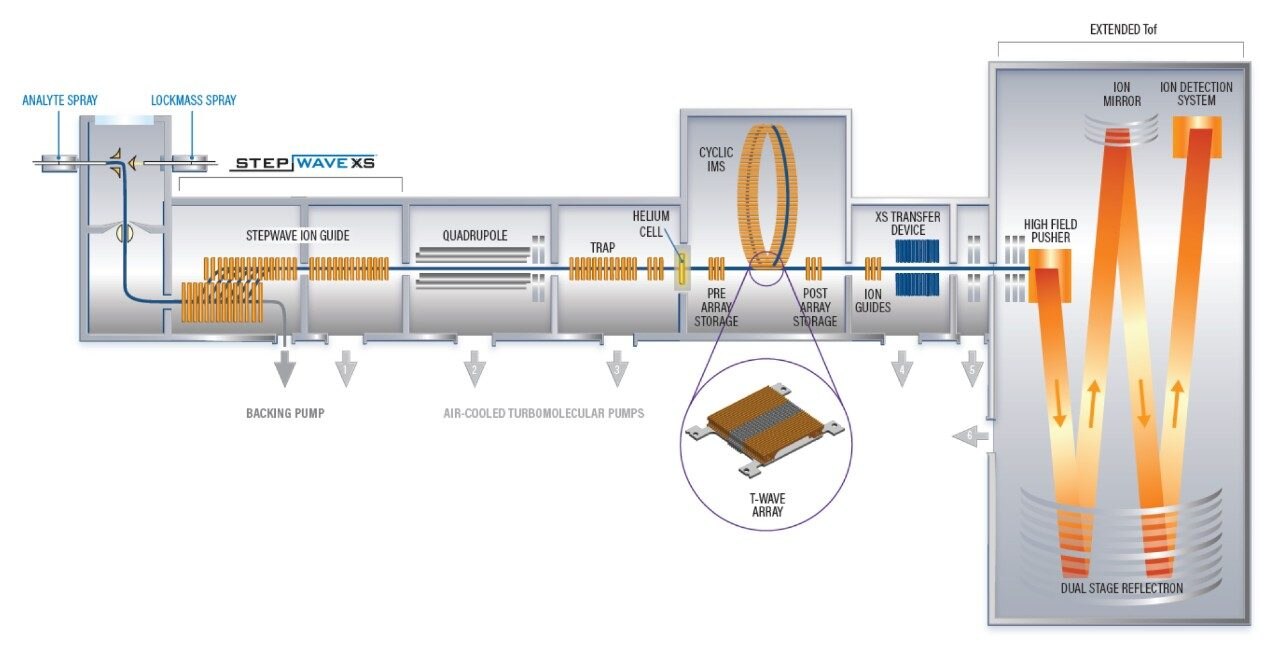
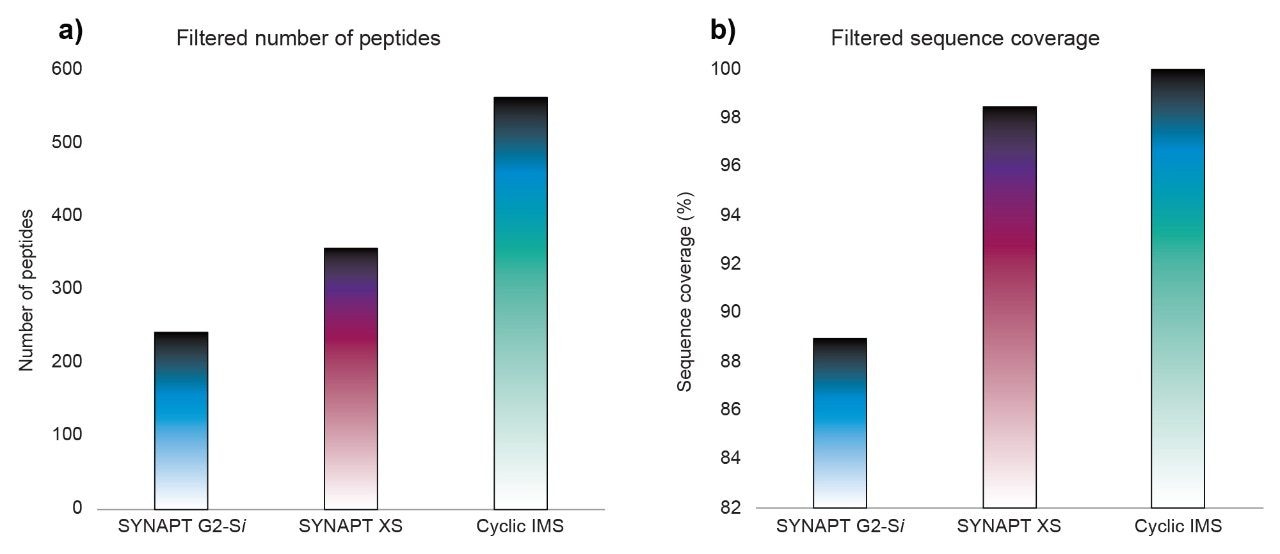
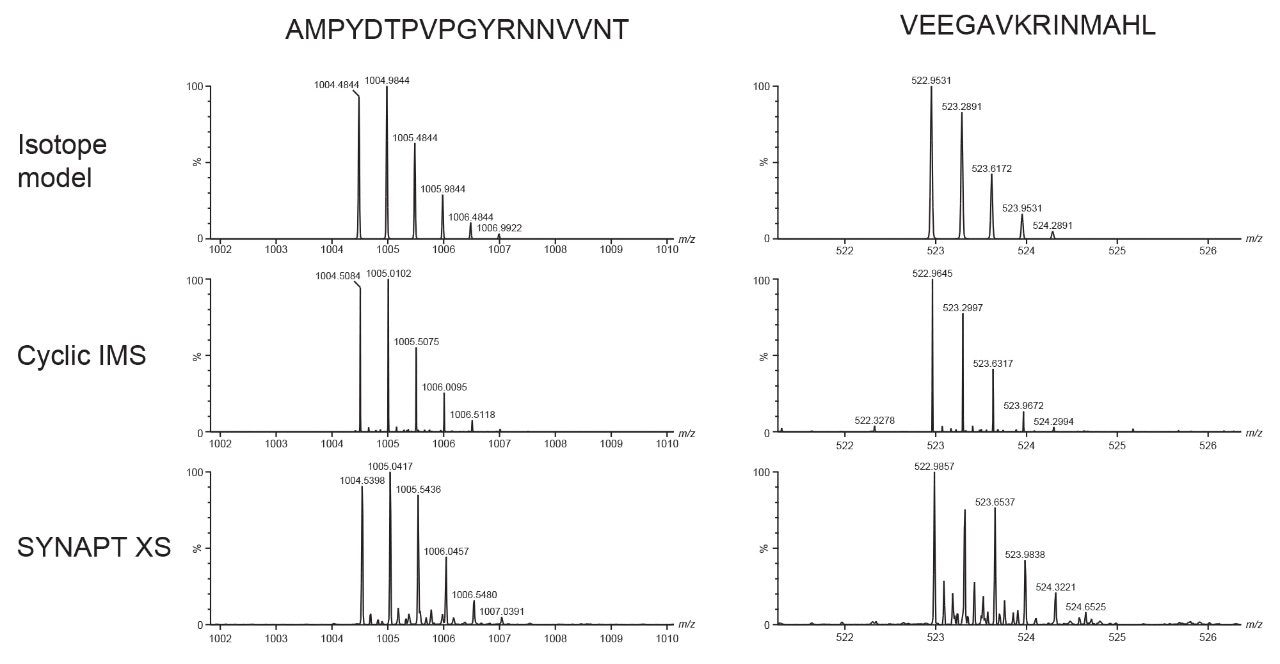
Characterization of higher order structure in proteins and protein complexes is integral for understanding the function and mechanisms of protein action and is essential to the evaluation and development of therapeutic biomolecules. Hydrogen deuterium exchange (HDX) facilitates localization of binding sites and regions of conformational change and provides information regarding flexibility and protein dynamics. HDX complements high spatial resolution techniques such as NMR, cryo-EM, and X-ray crystallography and results can typically be obtained with significantly higher throughput. Hence, HDX has been used as a cost-effective approach for epitope mapping screening and continued development regarding the throughput of the approach is expected to be widely beneficial.1
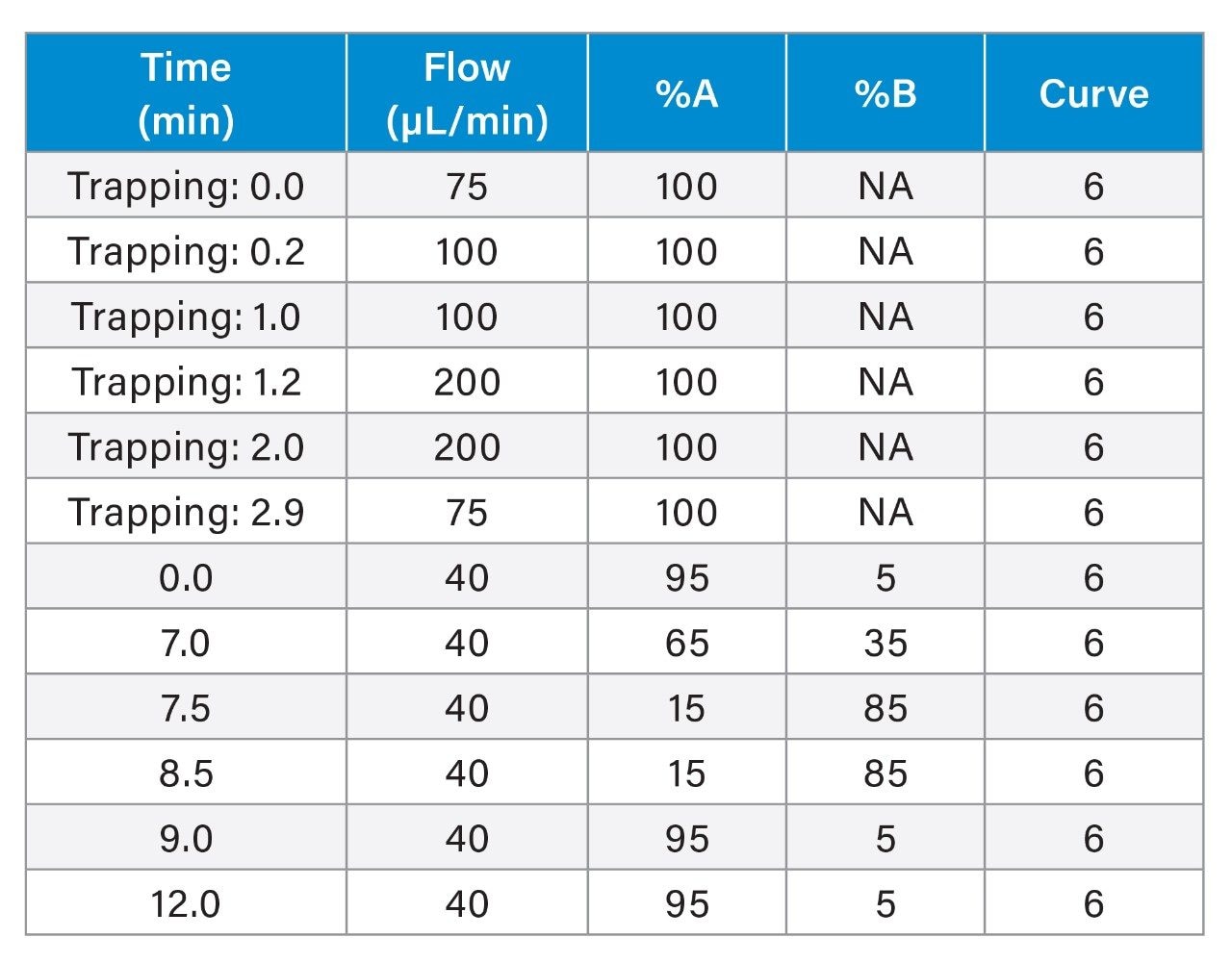
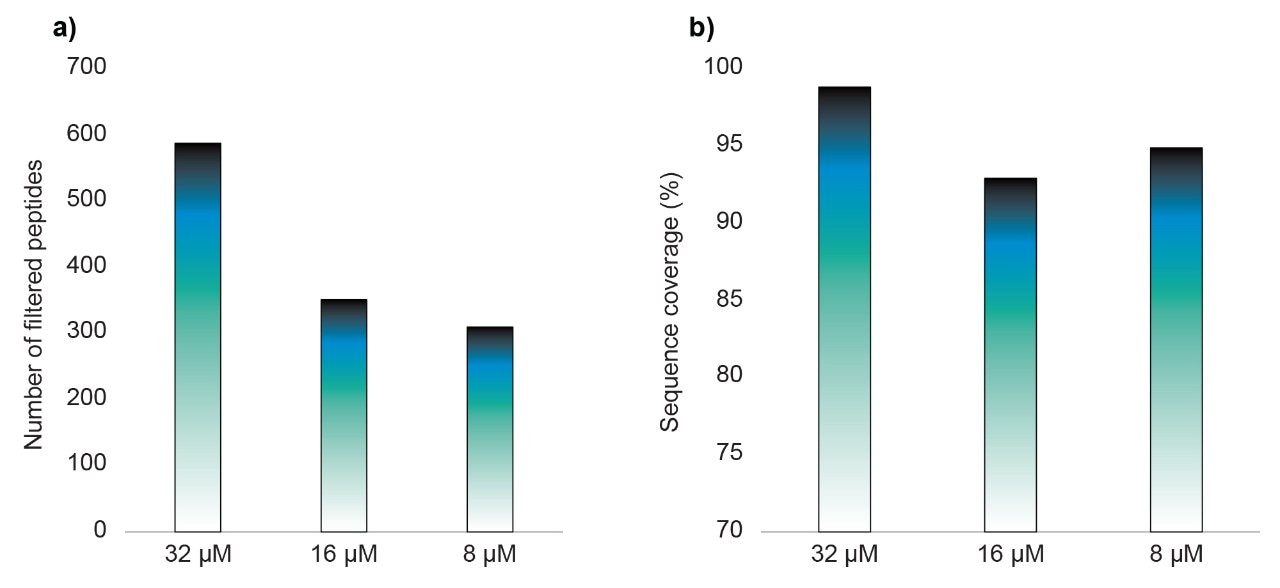
Amide hydrogen exchange occurs spontaneously in solution and can be exploited to monitor the hydrogen bonding protection that occurs along the backbone of proteins. Typical experimental protocols involve dilution of a stock protein into a deuterated buffer. Incubation in the deuterated buffer for variable time periods enables measurement of protein backbone kinetics in the seconds to hours timeframe. Measurement of deuterium uptake involves a low temperature, low pH quench followed by rapid pepsin digestion and LC-MS. Utilization of shorter chromatographic gradients yields higher deuterium retention but results in higher spectral complexity, which can be a challenge to in-silico database searches. Consequently, the quality of experimental data is typically a balance between protein size and chromatographic length. As the trend over recent years has been the study of larger protein complexes, often with tens of subunits, spectral complexity is increasingly a challenge.