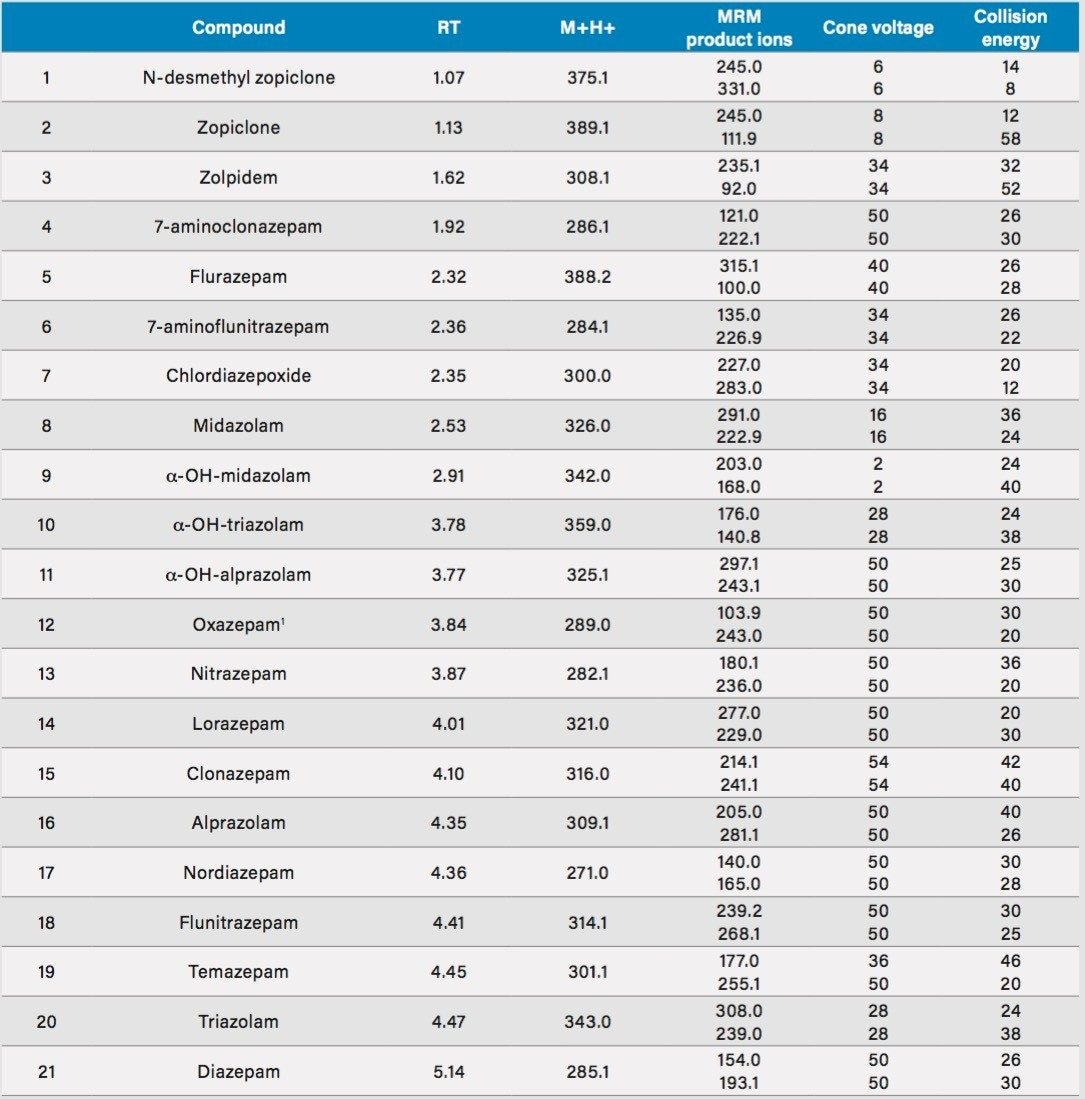
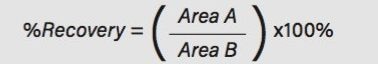Benzodiazepines are frequently prescribed drugs used for their sedative, anxiolytic, and hypnotic properties.1 They work by potentiating the inhibitory neurotransmitter gamma-amino butyric acid (GABA). Nationally, overdose deaths from benzodiazepines have risen 600% from under 1,600/year in 2001 to 8,000 in 2014, greater than any other drug class with the exception of heroin.2 So-called “Z-drugs” (zolpidem and zopiclone) are commonly used sleep aids that act in a similar manner to benzodiazepines.1 While the use of LC-MS/MS for benzodiazepine analysis has increased in recent years, many published techniques still rely on labor intensive liquid-liquid extraction techniques.3-5 Some of the drawbacks of these techniques include the need to process individual samples one by one, the use of toxic solvents, and the need to evaporate and reconstitute samples after extraction.
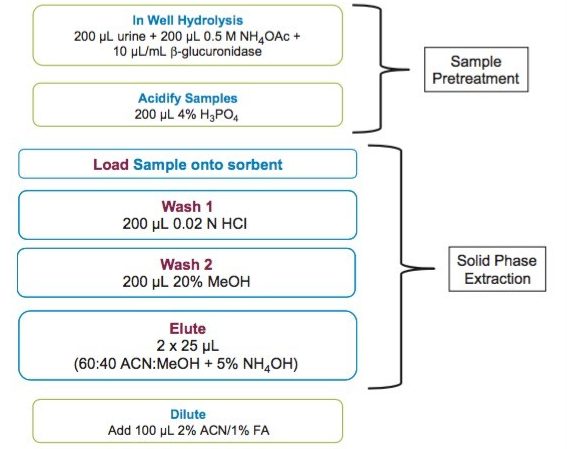
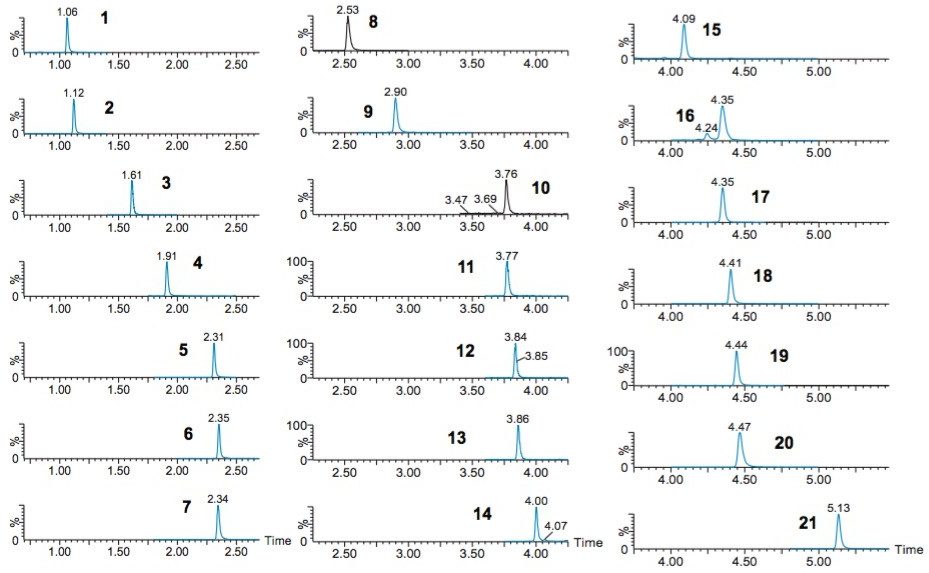
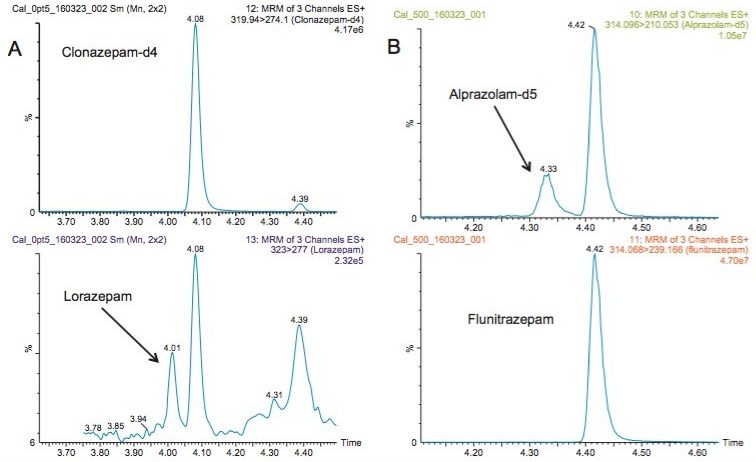
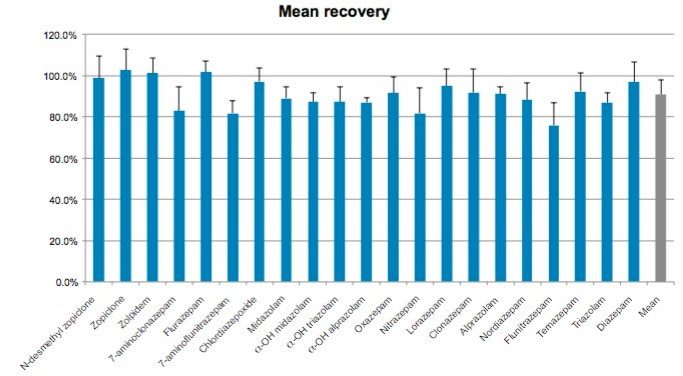
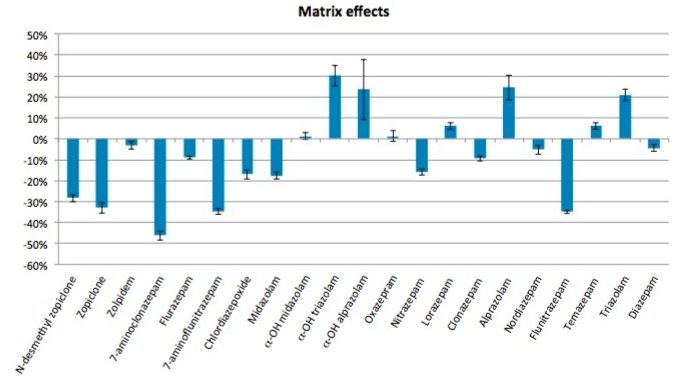
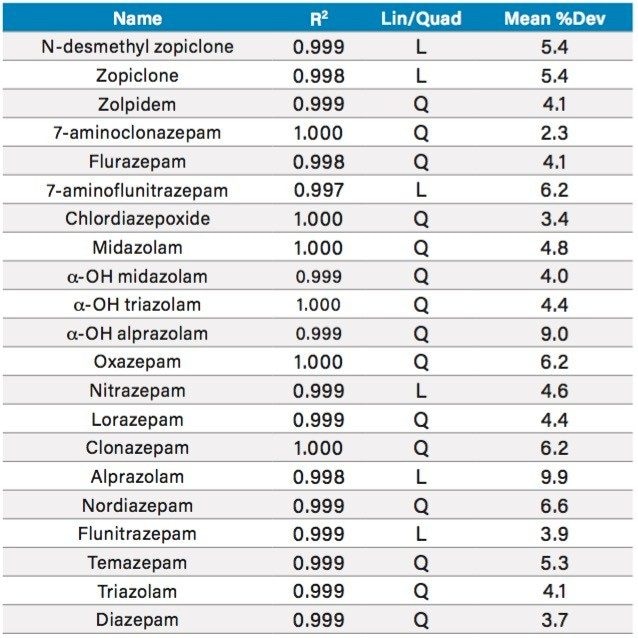
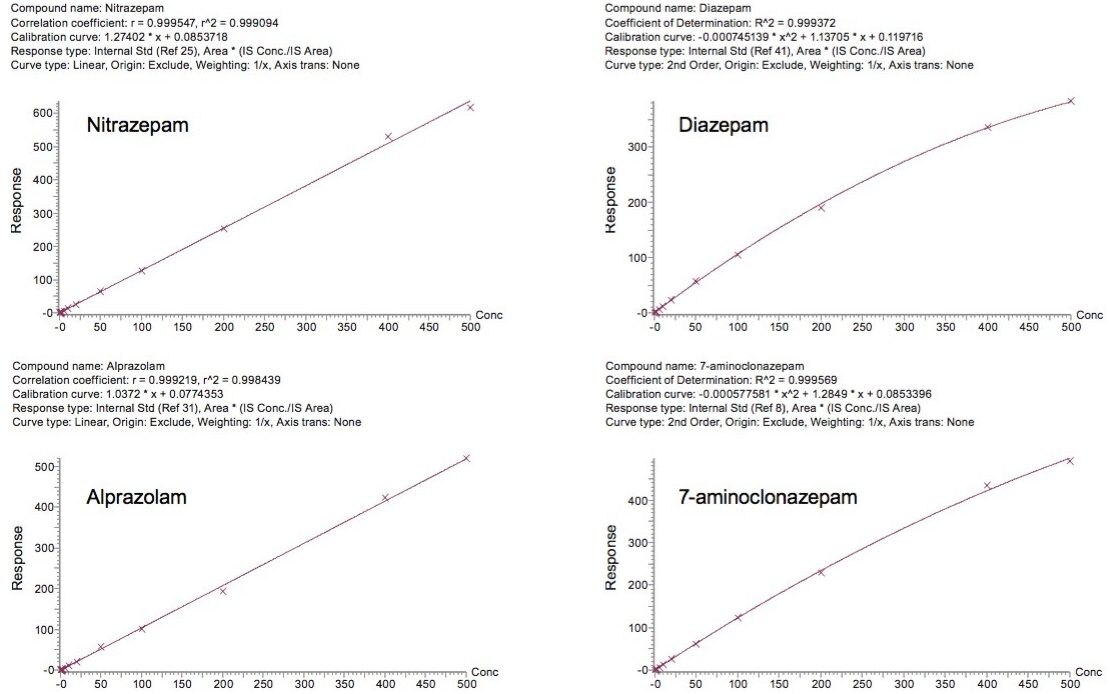
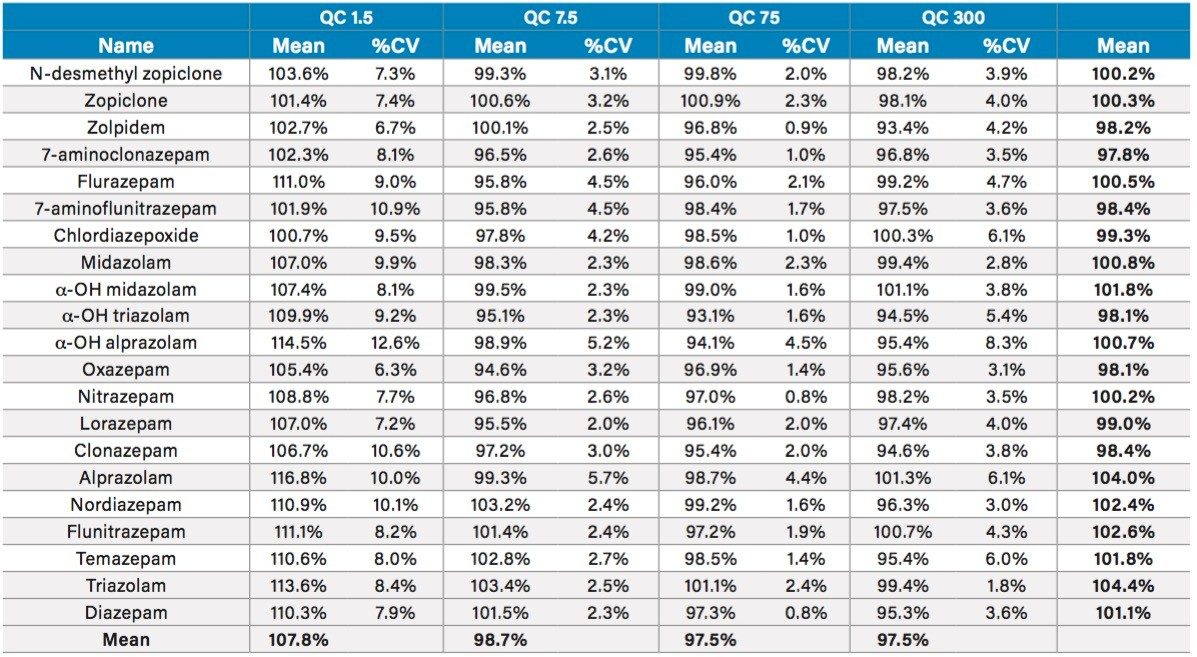
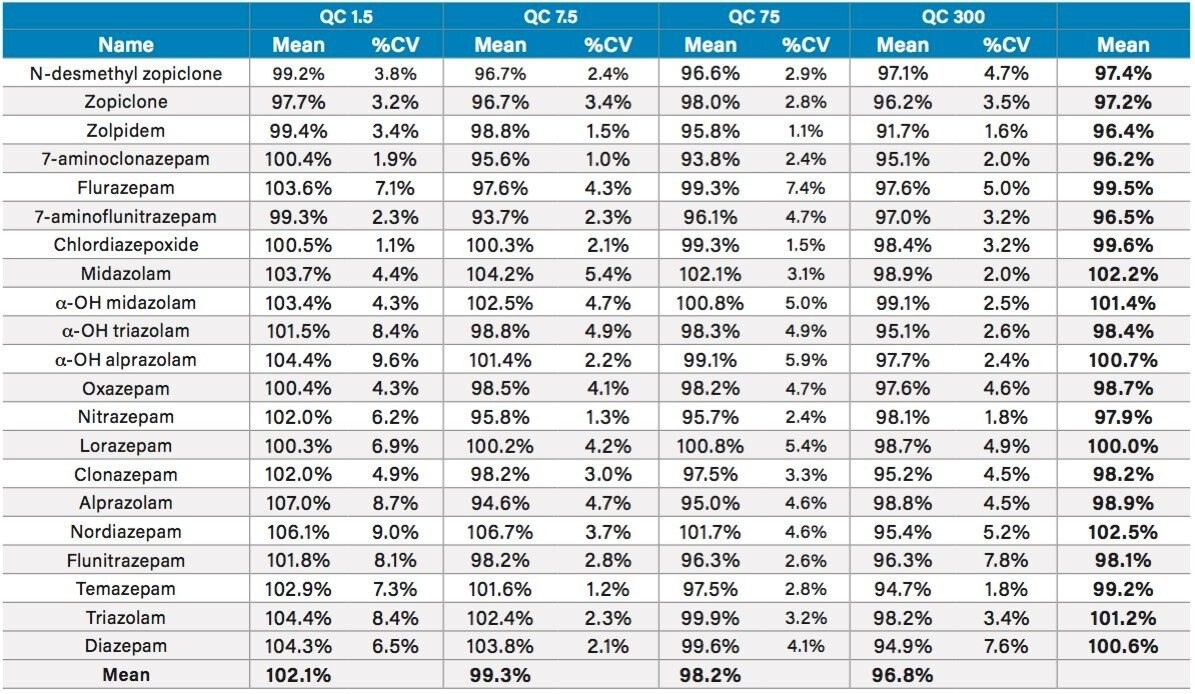
This application note details a sample preparation and LC-MS/MS analysis strategy for a comprehensive panel of benzodiazepines, metabolites, and Z-drugs for forensic toxicology use. Using an abbreviated, modified solid phase extraction (SPE) method, Waters Oasis MCX µElution Plates were used to rapidly extract this panel of drugs and metabolites from urine samples. All sample preparation steps, including enzymatic hydrolysis, were performed within the wells of the Oasis MCX µElution Plates, and the extraction method was simplified by eliminating conditioning and equilibration steps. This enabled a streamlined workflow that minimized sample transfer steps while still achieving excellent and reproducible quantitative results. Chromatographic separation was achieved using a CORTECS UPLC C18+ Column while a Xevo TQ-S micro Mass Spectrometer was used for detection. Extraction recovery was efficient, averaging 91%, and the use of the mixed-mode sorbent reduced matrix effects compared to reversed-phase SPE. The CORTECS UPLC C18+ Column enabled the baseline separation of all target analytes from internal standards with identical nominal masses. This eliminated the risk of chromatographic interference between the labeled internal standards and the native compounds. All within and between batch quality control samples had mean accuracies within 5% of nominal values.
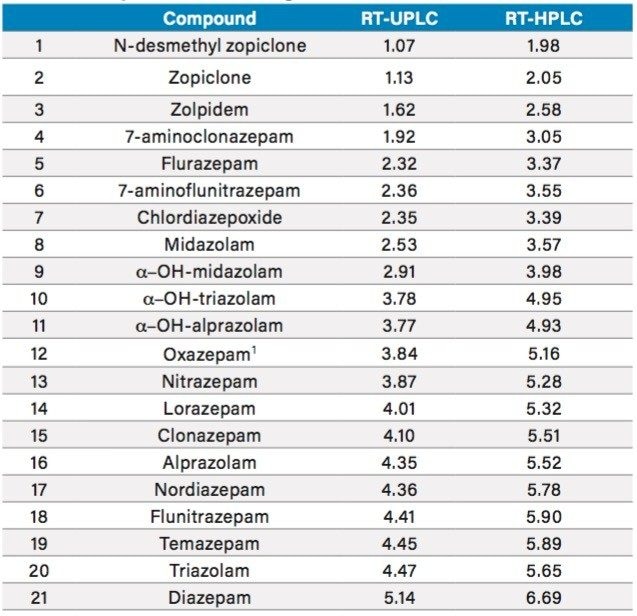
This method was also performed at HPLC scale using a CORTECS UPLC C18+ 2.7 µm Column (3.0 x 100 mm) (p/n 186007372). The same efficient separation was seen as with the 1.6 µm column (p/n 186007402), with backpressures that remained under 4000 psi and a separation time that was increased by only 30%.