Size-Exclusion Chromatography Method for Poly(A) Tail Analysis of mRNA
Abstract
The first in-class therapeutic messenger RNA (mRNA) vaccines were successfully developed against SARS-CoV-2 virus. Therapeutic mRNA molecules can also be used for protein replacement therapy or vaccination approaches in cancer treatment. Rapid advances of mRNA technology require development of analytical methods. Current mRNA methods include analysis of 5’ capping status, primary mRNA sequence, process impurities, and the 3’ poly(A) tail. In this application note, we describe a robust and simple method for poly(A) tail length measurements. The method utilizes the digestion of an mRNA molecule with RNase T1 to liberate poly(A) tail. The digestion products and poly(A) tail are then separated by size-exclusion chromatography (SEC). The poly(A) tail length is estimated using an SEC calibration constructed from nucleic acid standards.
Benefits
- Fast and robust method for analysis of poly(A) tail length
- Minimal sample preparation
- Simple SEC-UV method suitable for QC testing
Introduction
Therapeutic messenger RNA molecules are single stranded nucleic acids, typically 2–10 thousand nucleotides long. Therapeutic mRNA have an anatomy that consists of several distinct parts, including a modified 5’ cap, 3’ and 5’ untranslated regions (UTRs), the mRNA coding region or gene transcript sequence, and a poly(A) tail on the 3’ end (see Figure 1). The 5’ end cap minimizes RNA degradation while facilitating the translation process by latching on to ribosomes in the cell. The 3’ and 5’ untranslated regions directly impact translation and are often sequence optimized to achieve maximum RNA translation efficiency. The mRNA coding region encodes for the gene of interest (GOI). This is the part that gets translated into the desired protein, and it often contains modified nucleotides (typically N1-methylpseudouridine) to reduce rates of clearance and increase protein production. Finally, there is the poly(A) tail on the 3’ end, which plays a critical role in minimizing RNA degradation and maintaining in-vivo stability of the mRNA. The poly(A) tail portion in mRNA vaccines is typically 100–150 nucleotides (nt) long.
The success of the SARS-CoV-2 vaccines initiated significant investments into mRNA technology. Many clinical studies of mRNA therapies are in progress as well as mRNA vaccines targeting several pathogens and viruses. Recent advancements in mRNA technology have facilitated other promising applications too, such as cancer immunotherapy, in vivo CRISPR gene editing and other protein replacement therapies.
mRNA therapeutics are new modalities; they are large biopolymer molecules, which presents unique analytical challenges. New analytical methods are required to support mRNA research, as well as the development, manufacturing, and QC release of mRNA therapeutics and vaccines, including product characterization and impurities analysis. Because the direct analysis of large, intact nucleic acid molecules is difficult by liquid chromatography (LC) and mass spectrometry (MS) methods,1 intact mRNA molecules are often digested with specific enzymes, such as ribonuclease (RNase) T1.2 RNase T1 cleaves single stranded RNA at the 3’ end of guanosine nucleotides, leaving a phosphate group at the 3’ end of the product. Because guanosine is frequently present in the mRNA sequence, the RNase T1 digestion yields a mixture of relatively short oligonucleotides that can be separated and sequenced by LC MS/MS methods (3–5). Because the poly(A) tail portion of the mRNA molecule does not contain guanosine residues, an RNase T1 digest produces an oligonucleotide consisting predominantly of an oligo(A) sequence that is significantly longer than the other oligonucleotides present in the digest (see Figure 1). The length of this oligonucleotide is typically 100–150 nt long.
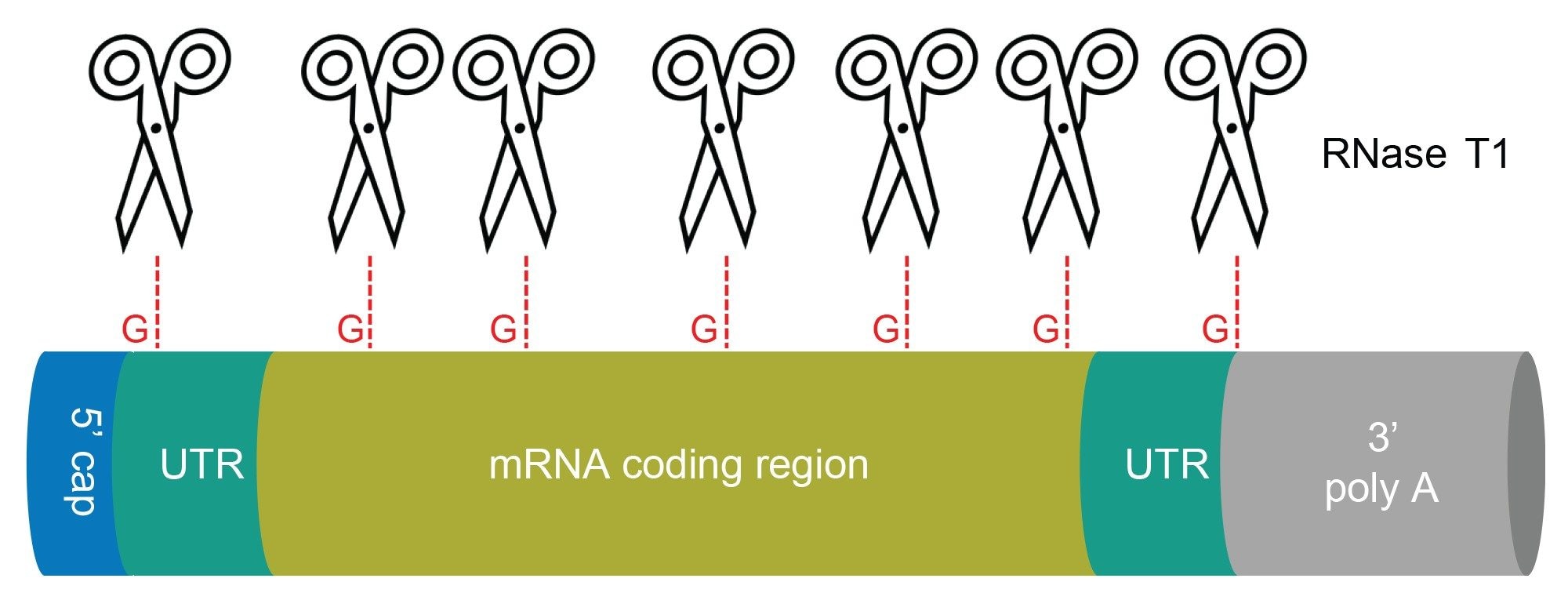 Figure 1. Schematic representation of mRNA structure. Scissors indicate putative cleavage sites of RNase T1. The digest produces short RNA oligonucleotides and liberates the 3’-end poly(A) tail.
Figure 1. Schematic representation of mRNA structure. Scissors indicate putative cleavage sites of RNase T1. The digest produces short RNA oligonucleotides and liberates the 3’-end poly(A) tail.
In this application note we describe a simple SEC method for determining the length of the poly(A) tail after intact mRNA digestion with RNase T1. The digestion products, 2–30 nt oligonucleotides and poly(A) tail oligonucleotide(s) are resolved by SEC. The average length of the mRNA poly(A) tail is obtained from the retention time of poly(A) oligonucleotide and a comparison to an SEC calibration curve prepared with adenosine ribonucleotide standards. This is a technique that is complementary to an Ion-Pair Reverse-Phase (IP RP) LC method, wherein it is possible to achieve n/n-1 oligonucleotides resolution for up to ~150 nt length species and to gain another viewpoint on poly(A) tail length heterogeneity. The IP RP LC method will be described in more detail in a subsequent application note 720007873.
Experimental
EPO mRNA standard was obtained from TriLink Biotechnologies, part number L-7209. EPO mRNA contains Cap 1 5’ structure, it is polyadenylated, and contains modified 5-methoxyuridine throughout the sequence. Fluc-beta mRNA sample was obtained from AmpTec, part number M1436/1000-C1-A120-NM-P0. The mRNA is polyadenylated, and contains unmodified uridines in the sequence. 1 mg/ mL solution of mRNA samples were digested with RNase T1 (Thermo Fisher, part number EN0542, 1000 U/µL) using the following protocol:
50 µL of 1 mg/mL mRNA was mixed with 10 µL of rCutSmart 10x buffer (New England Biolabs, part number B6004S) and 2 µL of RNase T1 was added. The sample was digested for 30 minutes at 37 °C. 1 µL of Quick CIP enzyme (New England Biolabs, part number M0525S, 5000 µ/mL) was added to the sample after the digestion to remove 3’ phosphate groups from the oligonucleotides. Dephosphorylation was performed for 30 minutes at room temperature. 1 µL of the resulting digest was injected onto the SEC column or analyzed by the Ion-Pair Reverse-Phase LC method.
SEC Conditions
|
LC system: |
ACQUITY™ Premier UPLC™ (with SM-FTN and QSM) |
|
Detection: |
PDA, Ti 5 µL cell, 260 nm |
|
Column: |
ACQUITY Premier Protein SEC 250 Å, 4.6 x 150 mm, 1.7 µm, (p/n: 186009963) |
|
Column temperature: |
25 °C |
|
Sample temperature: |
10 °C |
|
Injection volume: |
1.0 µL (sample) |
|
Flow rate: |
0.2 mL/min |
|
Mobile phase A: |
0.1 M phosphate buffer, pH 8, filtered through 0.1 µm filter |
|
Mobile phase B: |
30:70 methanol:water used for column storage |
|
Chromatography software: |
Empower™ v 3.0 |
IP RP LC conditions
|
LC system: |
ACQUITY Premier UPLC (with SM-FTN and QSM) |
|
Detection: |
PDA, Ti 5 µL cell, 260 nm |
|
Column: |
ACQUITY Premier Oligonucleotide BEH C18 300 Å, 2.1 x 150 mm, 1.7 µm, (p/n: 186010541) |
|
Column temperature: |
60 °C |
|
Sample temperature: |
10 °C |
|
Injection volume: |
1.0 µL (sample) |
|
Flow rate: |
0.3 mL/min |
|
Mobile phase A: |
100 mM octylammonium acetate (OAA) in 40% acetonitrile and 1% hexafluoroisopropanol (HFIP), v:v |
|
Mobile phase B: |
100 mM OAA in 90% acetonitrile and 1% HFIP |
|
Mobile phase D: |
100% acetonitrile |
|
Gradient: |
From 46% A, 54% B, and 0% D to 28% A, 62% B, and 10% D in 40 min |
|
Chromatography software: |
Empower v 3.0 |
Results and Discussion
SEC Size-Exclusion Chromatography is a method of separating molecules according to their hydrodynamic size. In the first step of this work, we screened Waters™ 150 x 4.6 mm SEC Column packed with 1.7 or 2.5 µm sorbents for their ability to separate 15–150 nt oligonucleotides. An ACQUITY UPLC BEH SEC 1.7 µm 125 Å Column was found to be suitable for separating 2–25 nt oligonucleotides. An ACQUITY Premier Protein SEC 250 Å 1.7 µm Column is suitable for 20–150 nt oligonucleotides, while an ACQUITY BEH SEC 450 Å 2.5 µm Column provides useful separation for 50–4000 nt nucleic acids (data not shown).
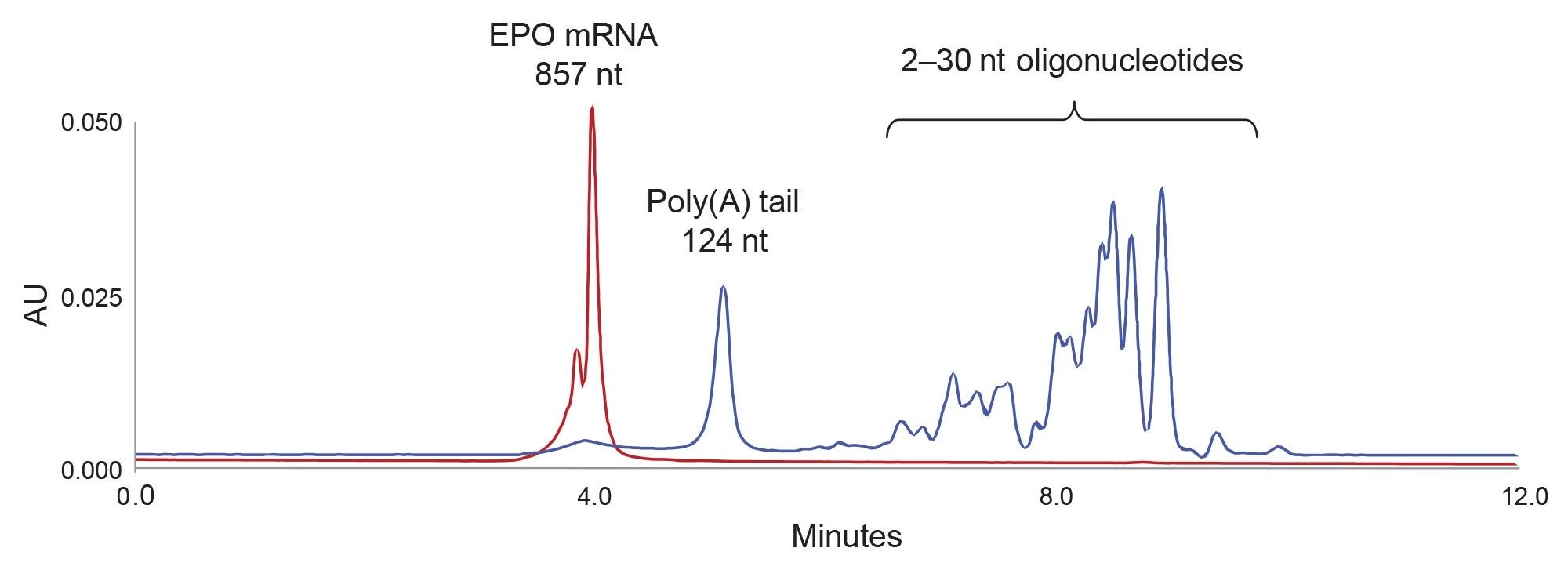 Figure 2. 857 nt EPO mRNA (red chromatogram) was digested with RNase T1. The digestion products, poly(A) tail and 2–30 nt short oligonucleotides (blue chromatogram) are separated with an ACQUITY Premier Protein SEC 250 Å, 4.6 x 150 mm, 1.7 µm Column.
Figure 2. 857 nt EPO mRNA (red chromatogram) was digested with RNase T1. The digestion products, poly(A) tail and 2–30 nt short oligonucleotides (blue chromatogram) are separated with an ACQUITY Premier Protein SEC 250 Å, 4.6 x 150 mm, 1.7 µm Column.
Based on this preliminary study, we selected the ACQUITY Premier Protein SEC 250 Å 1.7 µm Column for further experiments. Figure 2 shows that this column provides an excellent separation of undigested mRNA from the mRNA RNase T1 digestion products: namely the liberated poly(A) tail from the short oligonucleotides. The length of poly(A) tail assignment is shown in Figure 3 based on a calibration with the oligoribonucleotide adenosine standards.
The calibration trend logN = a × tr + b was linear for oligonucleotides in the 30–150 nt range. N is number of nucleotides, tr is retention time, and a and b are linear regression constants. This calibration was used to calculate the length of RNA poly(A) tail from the observed SEC peak retention time.
Figure 3b illustrates the SEC column calibration with synthetic RNA oligo(A) standards. 30, 40, and 60 nt samples show high purity, while the 50 nt oligo(A) RNA peak is significantly more contaminated with the truncated synthetic impurities. As expected, a number of synthetic impurities was detected in the 100 nt standard. The estimated purity of the 100 nt oligonucleotide (based on the SEC analysis) was ~ 9% for the full-length product.
When compared to oligo(A) standards (Figure 3b) one may notice an apparent poly(A) tail peak fronting and broadening (Figure 3a). This is because the poly(A) tail peak is not homogeneous; it consists of multiple closely related poly(A) tail species. An SEC method is not capable of resolving N-x and N+x poly(A) tail variants. However, the SEC UV method does provide a simple estimation for the length of the dominant oligonucleotide present in the poly(A) tail peak. If information about the heterogeneity of the poly(A) tail species is required, one can use a high resolution Ion-Pair Reverse-Phase LC method capable of resolving N and N-x species up to 150 nt oligonucleotides. Moreover, the high resolution IP-RP method can be optimized to be couple to mass spectrometry.
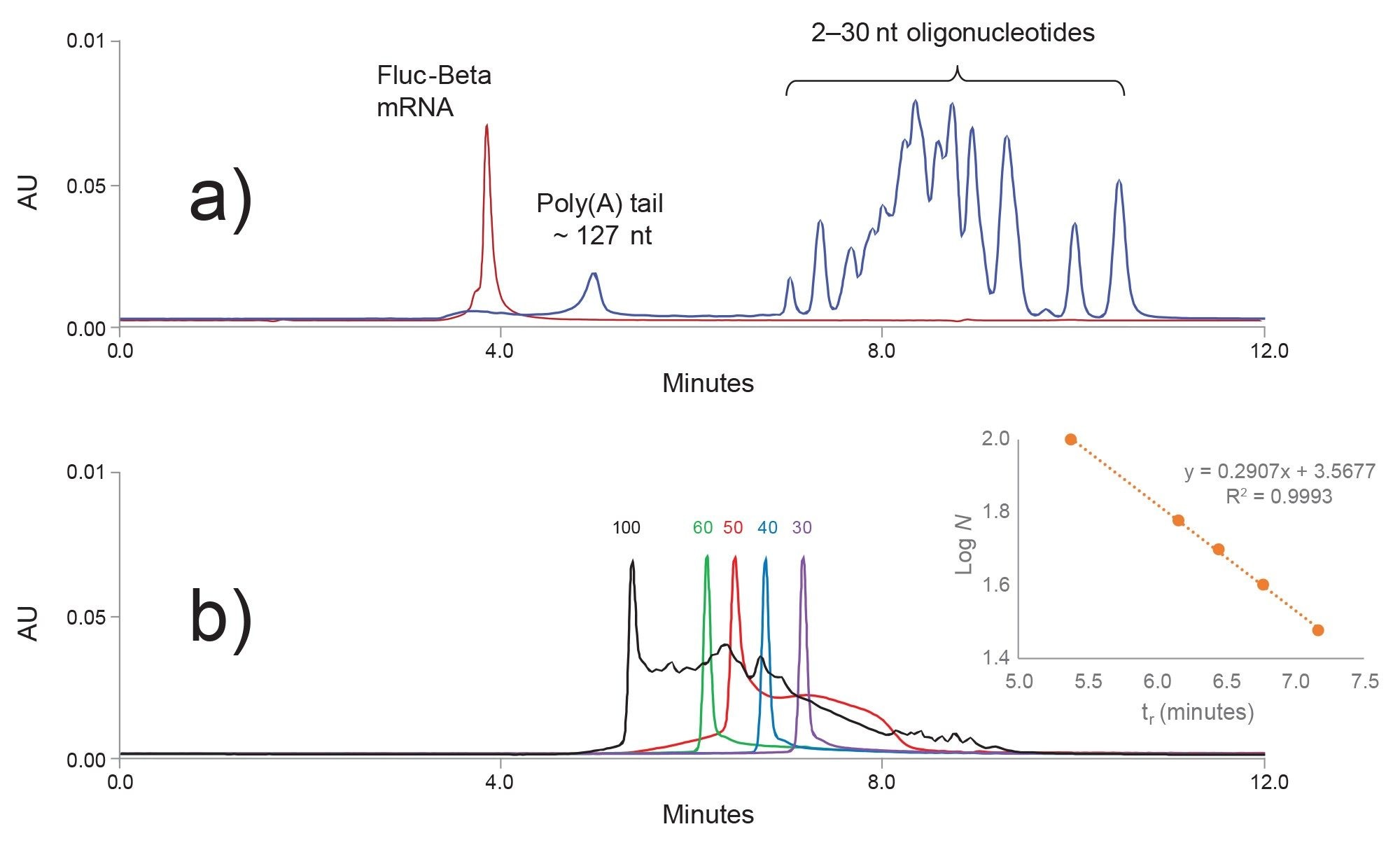 Figure 3. a) 1970 nt Fluc-beta mRNA (red chromatogram) was digested with RNase T1. The digestion products, poly(A) tail and 2–30 nt short oligonucleotides (blue chromatogram) were separated with an ACQUITY Premier Protein SEC 250 Å, 4.6 x 150 mm, 1.7 µm Column. b) SEC calibration with synthetic 30, 40 , 50, 60, and 100 nt oligoribonucleotide(A) standards. Linear calibration is shown in the inset. An extrapolation of the calibration curve is used to approximate the length of the even larger poly A tail.
Figure 3. a) 1970 nt Fluc-beta mRNA (red chromatogram) was digested with RNase T1. The digestion products, poly(A) tail and 2–30 nt short oligonucleotides (blue chromatogram) were separated with an ACQUITY Premier Protein SEC 250 Å, 4.6 x 150 mm, 1.7 µm Column. b) SEC calibration with synthetic 30, 40 , 50, 60, and 100 nt oligoribonucleotide(A) standards. Linear calibration is shown in the inset. An extrapolation of the calibration curve is used to approximate the length of the even larger poly A tail.
High-Resolution IP RP LC UV investigation of poly(A) tail heterogeneity
An efficient IP RP LC separation of 100–150 nt oligonucleotides requires columns packed with sub-two micron particles (6, 7), and a strong ion-pairing mobile phase, such is 100 mM octylammonium acetate (3). The separation of the 100 nt oligo(A) RNA synthetic standard and the poly(A) tail liberated from an EPO mRNA digest with RNase T1 is shown in Figure 4. Partial separation of n/n-x species is achieved in IP RP LC for oligonucleotides up to 150 nt. The poly(A) tail consists of 10–15 oligonucleotides centered around the most abundant peak with an estimated length of 124 nt. This particular IP RP LC mobile phase is not compatible with MS detection, the length of 124 nt peak was assigned with poly(A) RNA synthetic standards. This exact IP PR LC method will be described in further detail in the subsequent application note.
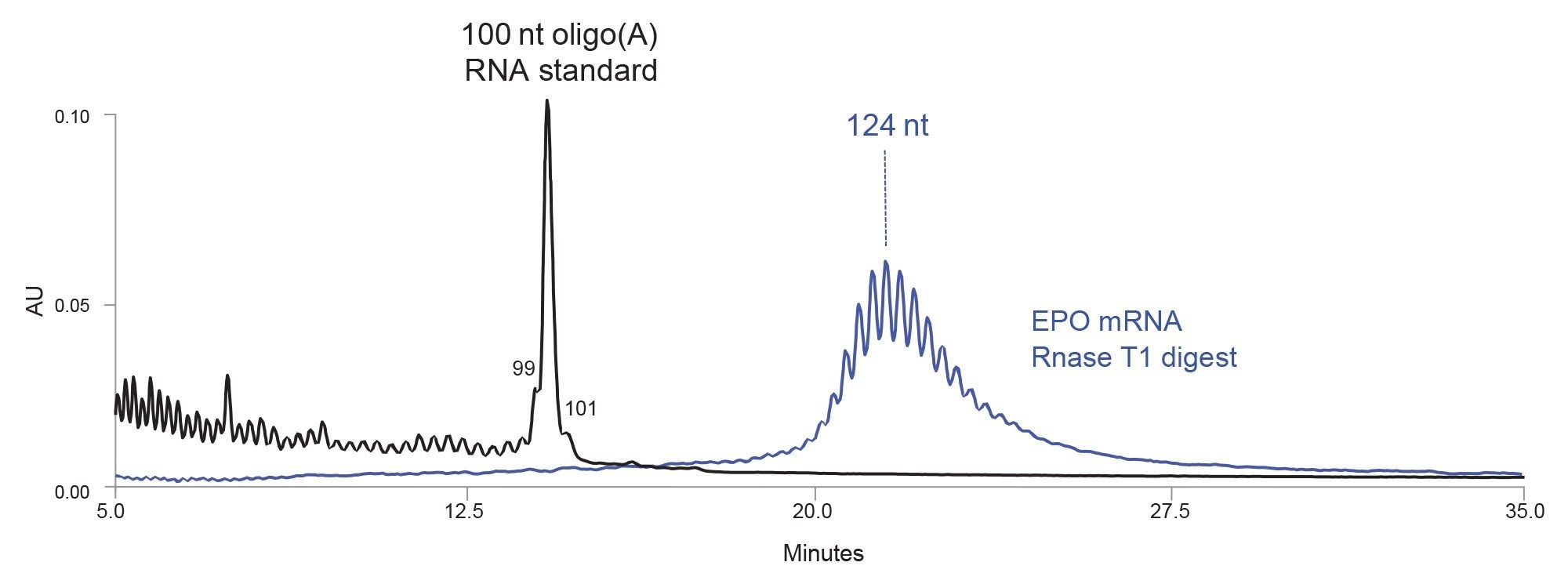 Figure 4. Separation of 100 nt oligo(A) synthetic RNA oligonucleotide standard (black chromatogram) and EPO mRNA digest with RNase T1 (blue chromatogram) with a high resolution IP RP LC method using an ACQUITY Premier Oligonucleotide BEH 300 Å 1.7µm Column. The most abundant peak amidst poly(A) tail signal is estimated to be 124 nt long.
Figure 4. Separation of 100 nt oligo(A) synthetic RNA oligonucleotide standard (black chromatogram) and EPO mRNA digest with RNase T1 (blue chromatogram) with a high resolution IP RP LC method using an ACQUITY Premier Oligonucleotide BEH 300 Å 1.7µm Column. The most abundant peak amidst poly(A) tail signal is estimated to be 124 nt long.
Conclusion
An SEC-UV method was developed for simple and robust analysis of mRNA poly(A) tail length. The ACQUITY Premier Protein SEC 250 Å Column with its 250 Å pore size was found to be most suitable for the analysis. SEC calibration with oligoribonucleotide(A) synthetic standards was linear in the range of 30–150 nt length. A second method, IP RP LC UV, was found to also be suitable for resolving long oligonucleotides up to 150 nt. This method uses an ACQUITY Premier Oligonucleotide BEH C18 300 Å Column as a potential quality control assay on poly(A) tail heterogeneity, and it too does not strictly require MS detection. Synthetic oligoribonucleotide(A) standards are used to assign poly(A) tail length and heterogeneity. In both instances, columns with MaxPeak™ High Performance Surfaces were employed to ensure robust recoveries of oligonucleotide species (8–11).
References
- M. Packer, D. Gyawali, R. Yerabolu, J. Schariter, and P. White, Nat. Commun., 12, 6777 (2021). DOI: 10.1038/s41467-021-26926-0.
- T. Jiang, N. Yu, J. Kim, J.R. Murgo, M. Kissai, K. Ravichandran, E.J. Miracco, V. Presnyak, and S. Hua, Anal. Chem., 91, 8500-8506 (2019). DOI: 10.1021/acs.analchem.9b01664.
- M. Donegan, J.M. Nguyen, and M. Gilar, J. Chromatogr. A, 1666, 462860 (2022). DOI: 10.1016/j.chroma.2022.462860.
- A. Goyon, P. Yehl, and K. Zhang, J. Pharm. Biomed. Anal., 182, 113105 (2020). DOI: 10.1016/j.jpba.2020.113105.
- V.B. Ivleva, Y.Q. Yu, and M. Gilar, Rapid Commun Mass Spectrom, 24, 2631–2640 (2010). DOI: 10.1002/rcm.4683.
- M. Gilar and U.D. Neue, J. Chromatogr. A, 1169, 139–150 (2007). DOI: 10.1016/j.chroma.2007.09.005.
- M. Gilar, K.J. Fountain, Y. Budman, U.D. Neue, K.R. Yardley, P.D. Rainville, R.J. Russell, 2nd, and J.C. Gebler, J. Chromatogr. A, 958, 167–182 (2002).
- G.J. Guimaraes, J.M. Sutton, M. Gilar, M. Donegan, and M.G. Bartlett, J. Pharm. Biomed. Anal., 208, 114439 (2022). DOI: 10.1016/j.jpba.2021.114439.
- J.M. Nguyen, M. Gilar, B. Koshel, M. Donegan, J. MacLean, Z. Li, and M.A. Lauber, Bioanalysis, (2021). DOI: 10.4155/bio-2021-0115.
- M. Gilar, M. DeLano, and F. Gritti, J. Chromatogr. A, 1650, 462247 (2021). DOI: 10.1016/j.chroma.2021.462247.
- M. DeLano, T.H. Walter, M.A. Lauber, M. Gilar, M.C. Jung, J.M. Nguyen, C. Boissel, A.V. Patel, A. Bates-Harrison, and K.D. Wyndham, Anal. Chem., 93, 5773–5781 (2021). DOI: 10.1021/acs.analchem.0c05203.
720007853, February 2023