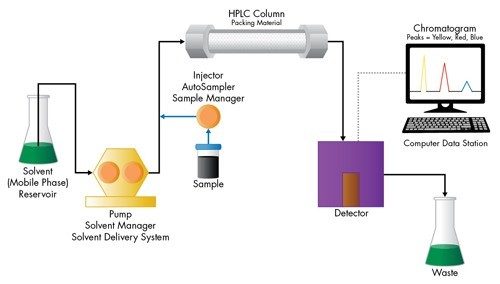
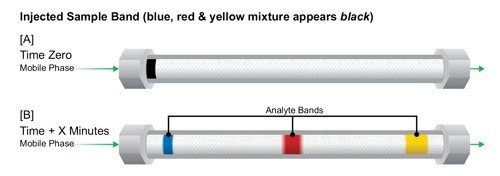
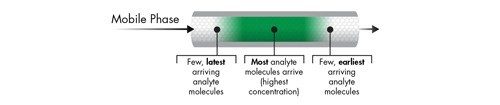
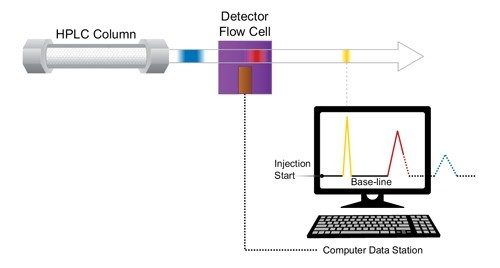
The Impact of Detector Settings on Extra-column Band Spreading
In addition to fluidic-based band spreading contributions of the instrument, digital detector settings related to acquisition speed and filter constant also influence the chromatographic result. This is especially important for UPLC applications, since peak widths are often very narrow [1–2 seconds wide], and analysis time can be very short. When setting the acquisition speed of the detector, the selection should be based upon acquiring sufficient data points across a peak to properly represent the peak. Setting the detector rate too high will negatively impact the signal-to-noise ratio causing the baseline noise to increase without an increase in the signal height of the analyte of interest. Conversely, setting the detector acquisition rate too slow will result in inadequate data points across the peak, thus reducing the observed chromatographic efficiency and compromising the ability to reproducibly perform quantitation. In addition, a time constant [digital filter] is used to smooth out data points to optimize signal-to-noise and can be used conjointly with or independent from, the acquisition speed.
Detector settings become increasingly more important as the width of the analyte band narrows. This requires an acquisition speed that is not only fast enough to digitally create a peak of that speed, but also to properly render the high resolution separation achieved in the UPLC column, if closely eluting peaks exist.
At slower flow rates when peaks are more disperse in time, these settings are more forgiving. However, as the peaks become narrower and the analysis more rapid [as in UPLC separations], a judicious setting of acquisition speed and time constant must be maintained [Figure 13]. When using UPLC Technology for ‘real-life’ applications, it is wise to select the detector data acquisition rate [Hz] that accurately captures the peak shape of the narrowest peak, and then apply a filter time constant [seconds] that gives optimal signal-to-noise and resolution for the analysis.
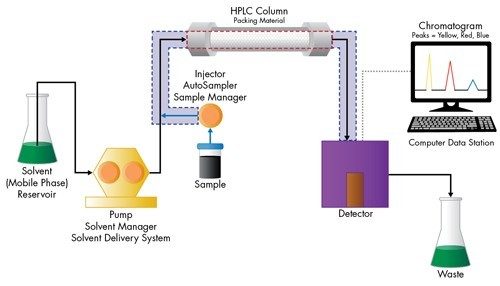
![Figure 8: As a statistical function, plate count can be thought of as the population variance [σ2], which is a summation of both extra-column and column variances. The population variance [σ2] measures the narrowness of a Gaussian peak [σ] relative to its elution volume.](/content/dam/waters/en/figures/primers/uplc/primer_uplc_08.jpg.82.resize/img.jpg)
![Figure 9: Impact of system band spreading [column and extra-column] on peak shape.](/content/dam/waters/en/figures/primers/uplc/primer_uplc_09.jpg.82.resize/img.jpg)
![Figure 11: The significant impact of instrument band spreading on column performance. The same column was run on an ACQUITY UPLC System and a conventional HPLC system. [ACQUITY UPLC BEH C18 2.1 x 50 mm, 1.7 µm column; flow rate = 0.4 mL/min.]](/content/dam/waters/en/figures/primers/uplc/primer_uplc_11.jpg.82.resize/img.jpg)
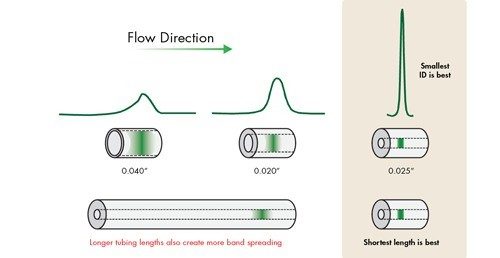
![Impact of acquisition speed [Hz] and time constant[s] on peak formation](/content/dam/waters/en/figures/primers/uplc/primer_uplc_13.jpg.82.resize/img.jpg)