Personen, die sich mit der Polymercharakterisierung durch chromatographische Techniken beschäftigen, verwenden nicht ausschließlich die GPC zur Analyse ihrer Proben. Häufig müssen wir Verfahren der Flüssigchromatographie durch Adsorption oder Verteilungschromatographie einsetzen, um die benötigten Informationen zu erhalten.
Herkömmliche Umkehrphasen- und gelegentlich auch Normalphasen-Techniken werden z. B. zur quantitativen Bestimmung von Polymeradditiven verwendet. Das Ermitteln der Molekulargewichtsverteilung Ihrer Polymerprobe kann nur ein Teil der Charakterisierung sein. Was ist mit den Additiven, die dem Polymer beigemischt werden, um eine Stabilisierung oder Verbesserung der Verarbeitung zu erreichen? Sie können sogar wichtiger sein als das Polymer selbst. Wir müssen darüber nachdenken, die richtigen UV-Stabilisatoren und Antioxidantien zum Schutz vor dem Abbau, Weichmacher zur Verbesserung der Flexibilität, Antistatika für Polyolefine, Flammschutzmittel, Beschleuniger zur Verbesserung der Vernetzung (oder Härtung) usw. zu verwenden.
Wir haben uns intensiv mit Polymeradditiven beschäftigt und Sie können einige unserer veröffentlichten Arbeiten im Journal of Liquid Chromatography, Band 14 #3, (1991), und Band 16, #7 (1993), finden.
Wie analysieren wir Polymeradditive? Zuerst müssen wir darüber nachdenken, was wir erreichen möchten. Müssen wir wissen, ob die korrekten Mengen jedes Additivs in der Formulierung vorhanden sind? Versuchen wir, ein Konkurrenzmaterial zu „deformulieren“? Müssen wir das Additivpaket aus der Polymermatrix extrahieren? Die Chancen stehen gut, dass die Antworten auf diese Fragen „Ja“ lauten. Die GPC-Analyse ist nicht der beste Weg, um vorhandene Zusatzstoffe in ihren Mengen zu trennen, zu identifizieren und zu quantifizieren. Die meisten Additive liegen in Größe und Molekulargewicht recht nahe beieinander, sodass wir sie mit HPLC trennen müssen. Eine einfache Gradiententechnik mit optionaler Flussprogrammierung funktioniert sehr gut, um viele verschiedene Arten von Additiven in kurzer Zeit zu trennen. Eine Gradientenanalyse besteht darin, die Zusammensetzung des Eluenten oder der mobilen Phase zu verändern, normalerweise von einem „schwachen“ Lösungsmittel zu einem „starken“ Lösungsmittel über einen gewissen Zeitraum. Diese Variation der Zusammensetzung erfolgt bei der additiven Analyse normalerweise auf lineare Weise. Da die Eluentenzusammensetzung während des Chromatographielaufs variiert wird, kann der Brechungsindex-Detektor nicht verwendet werden.
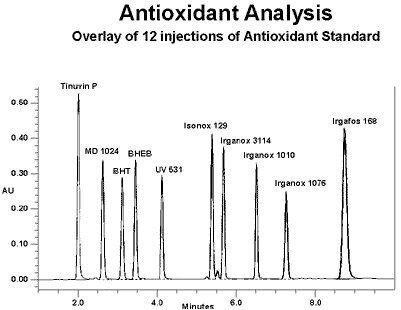
Die meisten Polymeradditive, mit denen wir zu tun haben, haben ein Chromophor, das ultraviolettes Licht absorbiert, so dass in erster Linie ein UV-Detektor verwendet wird. Wenn keine Chromophore vorhanden sind, kann ein Verdampfungslichtstreudetektor verwendet werden. Wir können die Flussrate auch während des Laufs ändern, wobei normalerweise der Fluss erhöht wird, damit die späteren eluierenden Komponenten schneller eluieren. Die für die Additivanalyse üblicherweise gewählte Säule ist eine Octadecylsilan- (C18) oder Octylsilan- (C8) Säule mit einer Länge von ca. 15 cm. Ein Beispiel für eine Umkehrphasengradiententrennung (mit Flussprogramm) einer Reihe von gebräuchlichen Antioxidantien und UV-Stabilisatoren mit 9 Überlagerung von 12 Injektionen wird hier gezeigt.



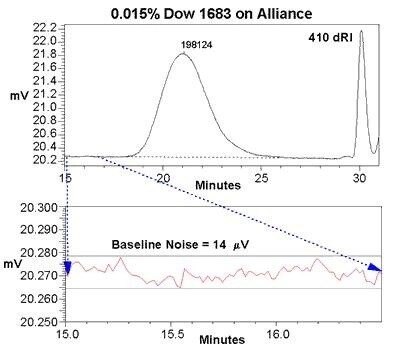
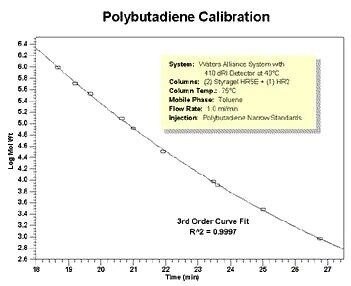




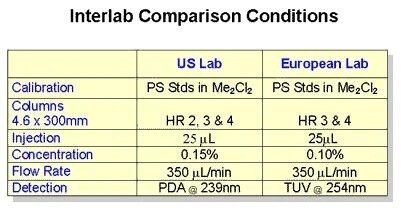








.jpg.82.resize/img.jpg)