不断发展的亚硝胺指导法规

2018年7月,欧洲药品管理局(EMA)报告称,由于受到已知致癌物二甲基亚硝胺(又称N-亚硝基二甲胺(NDMA))的污染,召回了一些含有活性药物成分(API)缬沙坦的产品。 这一行动引发了一连串的事件,导致多个地区当局发出类似的召回沙坦类产品的通知。 此外,在宣布发现NDMA之后,很快就有报告称发现了另一种N-亚硝胺--N-亚硝基二乙胺(NDEA)。
这些事件可以理解地触发了许多当局,包括欧盟委员会(EC),发起正式调查,旨在确定污染的根本原因。 欧盟委员会于2018年7月5日根据第2001/83/EC.号指令第31条启动了一项程序,概述了对控制和尽可能避免沙坦中N亚硝胺的期望。自那时起,与其他N-亚硝胺有关的进一步关注的报告不断出现,概述如下。
最初的第31条程序专门针对被称为 "沙坦类 "的药物,但即使在这一单一类别中,也有一个复杂的过程来确定每个沙坦类产品的安全限制。ICH M7中定义的原则取决于每种N-亚硝胺污染物的相对安全数据和每种沙坦类药物的日剂量,其范围从32mg/天到320mg/天。

有点争议的是,这些限制被定义为适用于2年的临时限制,此后对任何沙坦中的任何N-亚硝胺规定了0.03ppm的单一全面限制。 这代表着从科学的、基于安全的限值向 "合理可行的低 "方法(ALARP)的重大转变,使N-亚硝胺成为 "特例"。
与此同时,其他监管机构也采取了类似的行动,包括瑞士医疗机构、加拿大卫生部和其他众多机构。 美国FDA采取了某种不同的方法,要求报告检测到的任何级别的N-亚硝胺。 美国食品和药物管理局通过一系列具体的要求,在发给各个上市许可持有人的一般建议信中进一步透露了其想法。
- 除非原料药生产商根据指南(如ICH Q7)证明其工艺没有产生可检测亚硝胺的风险,否则应继续进行批量测试,以验证原料药中没有可检测亚硝胺。
这包括证明:
- 起始材料,包括供应商提供的中间体,没有可检测到的亚硝胺,或者可以清除少量的亚硝胺,以确保原料药不含有可检测到的数量,以及
- 在这个过程中使用的原材料,包括回收的溶剂和催化剂,不含有可检测的亚硝胺。
通过关注该过程中使用的起始材料、溶剂和催化剂,调查的范围被大大扩展。 这在很多方面都预示着接下来的事情。
2019年9月,与N-亚硝胺有关的情况发生了巨大的变化。 来自EMA、FDA和包括《纽约时报》在内的更广泛的公共媒体的报告,报道了在雷尼替丁中检测到NDMA,称由于其独特的结构,它可以降解,从分子本身产生NDMA。
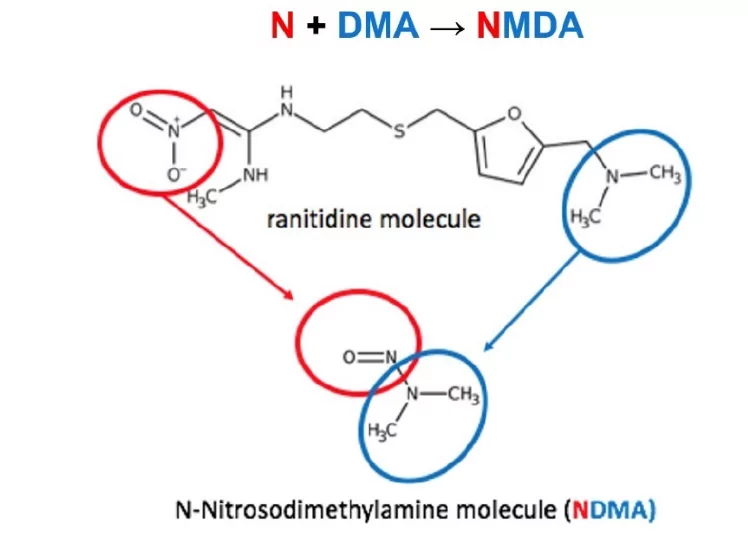
也许是由于这个原因,这引发了EMA引入了第二个范围更广的程序,即第5(3)条--为上市许可持有人提供关于亚硝胺的信息,要求对任何含有化学合成活性药物成分的人类医药产品中的亚硝胺杂质的风险进行评估。
这将调查范围从沙坦类药物扩大到了所有的合成分子,最初的最后期限为6个月,非常紧迫,不过后来延长到了2020年10月1日,然后再次延长到2021年3月。 伴随着一份问答文件,第5(3)条规定了以下要求。
在6个月内评估每种药品中存在亚硝胺的可能性。
- 确定评估的优先次序,从更有可能含有亚硝胺的药品开始
- 考虑到CHMP对沙坦类药物审查的结果
- 将风险评估的结果通知当局
- 测试有可能含有任何亚硝胺的产品
- 发现亚硝胺后立即向当局报告
- 为解决亚硝胺风险,申请对上市许可进行必要的修改
- 在3年内完成所有步骤,优先考虑高风险产品
评价工作分为2个步骤
步骤1 - 风险评估。上市许可持有人(MAH)应对其含有化学合成API的医药产品进行风险评估。要求MAH对产品进行优先排序,以便根据ICH M7和ICH Q9中规定的原则,确定对其产品进行评估的顺序。
风险评估结束后,MAH应通知相关主管部门。如果评估结果发现存在亚硝胺的风险,MAH应进入步骤2。
步骤2-确认性测试。如果在步骤1中进行的评估确定了存在亚硝胺的风险,应进行确认性测试。任何需要对生产过程进行的改变,都应在通知公布后的3年内完成。
如果测试确认存在亚硝胺杂质,无论检测到的数量是多少,MAHs都应立即通知相关部门。 最重要的是,没有提到具体的限制。
同时,加拿大卫生部、瑞士医疗机构和其他机构也提出了类似但不完全相同的要求,这使得对每个机构的答复变得更加复杂。 虽然值得欢迎,但3月份宣布推迟EMA第5(3)条的实施带来了进一步的挑战,因为并非所有其他机构都立即修改了他们的时间表。
在2020年7月的另一个转折中,EMA发布了他们最终的第5(3)条报告,不久之后还第三次修订了他们的相关问答文件。 其中重要的是以下变化。

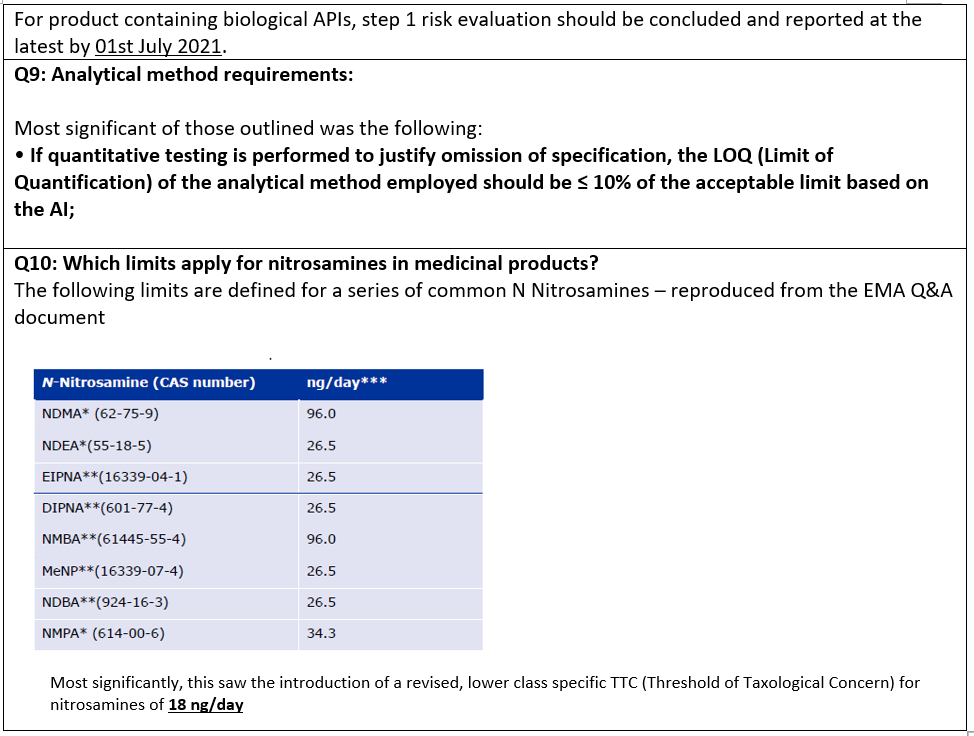
令人担忧的是,这个18纳克/天的通用限值,再加上检测药物产品允许摄入量(AI)10%的方法要求,将使一些药物产品的分析在技术上不可行。随着利用高性能专业仪器的分析方法使更低的检测限成为可能,监管当局是否会继续追逐更低的规格?这将是监管变化的终点吗?在这个阶段,不可能说,但不能排除。这些问题将在本系列的最后一篇博客中进一步研究。
相关帖子

为患者寻求更安全、更有针对性的疗法
我们的目标是通过扩大沃特世和惠悦技术的全球影响力和组合,创造新的价值。这两项技术以及我们深厚的科学专业知识将为我们的客户提供支持,使改变生命的疗法更容易获得。

沃特斯以直观简洁的方式突破了界限
我们相信我们已经创造了我们有史以来最直观简单的HPLC。它是有史以来第一个致力于满足质控实验室特定需求的系统,它将为我们的客户节省时间、金钱和压力。

透视医药:医药行业的变革管理
制药公司越来越重视新技术和创新,这有助于为患者提供质量更稳定、更安全的产品。