The Evolving Regulations Guiding Nitrosamines

In July 2018, the European Medicines Agency (EMA) reported the recall of a number of products containing the active pharmaceutical ingredient (API) valsartan due to contamination with a known carcinogen, dimethyl nitrosamine, also referred to as N-nitroso dimethylamine (NDMA). This action set off a chain of events that led to similar notifications of recalls for several sartan products across multiple regional authorities. Moreover, the announcement of the discovery of NDMA was quickly followed by reports of the detection of another N-Nitrosamine – N-Nitroso diethylamine (NDEA).
These events understandably triggered many authorities, including the European Commission (EC), to launch formal investigations aimed at identifying the root causes of the contamination. The EC initiated a procedure on 5 July 2018, pursuant to Article 31 of Directive 2001/83/EC., outlining the expectations for control and, where possible, avoidance of N Nitrosamines in sartans. Since then, reports of further concerns relating to other N-Nitrosamines have continued, as summarized below.
The initial Article 31 process was specific to the class of drugs referred to as “sartans”, but even within this single class there is a complex process to establish safety limits for each sartan based product. The principles defined within ICH M7 are dependent on the relative safety data of each N-Nitrosamine contaminant and the daily dose of each sartan, which can range from 32mg/day to 320mg/day.

Somewhat controversially, these were defined as interim limits applicable for a 2-year period, after which a single blanket limit of 0.03ppm was established for any N-Nitrosamine in any Sartan. This represents a significant shift away from scientific, safety-based limits, toward an “as low as reasonably practical” approach (ALARP), making N-Nitrosamines a ‘special case’.
At the same time, a similar action was taken by other regulatory authorities including Swiss Medic, Health Canada, and a plethora of others. The United States FDA took a somewhat different approach requesting that detection of any N-Nitrosamine at any level be reported. The FDA revealed further its thinking in a general advice letter sent to individual marketing authorization holders through a series of specific requirements:
- Batch testing to verify no detectable nitrosamine in the API should continue unless the API producer has demonstrated their process is not at risk for producing detectable nitrosamines in accordance with guidance (for example, ICH Q7).
This includes demonstrating that:
- starting materials, including vendor-supplied intermediates, have no detectable nitrosamines or such low amounts can be purged ensuring that the API contains no detectable amounts of nitrosamines, and
- raw materials used in the process, including recovered solvents and catalysts, contain no detectable amounts nitrosamines.
By also focusing on starting materials, solvents, and catalysts used in the process, the scope of investigations was significantly extended. This was, in many ways, a portent of what followed.
In September 2019 the situation relating to N-Nitrosamines changed dramatically. Reports from the EMA, FDA, and the wider public press, including the New York Times, reported the detection of NDMA in ranitidine, stating that due to its unique structure, it could degrade to generate NDMA from the molecule itself.
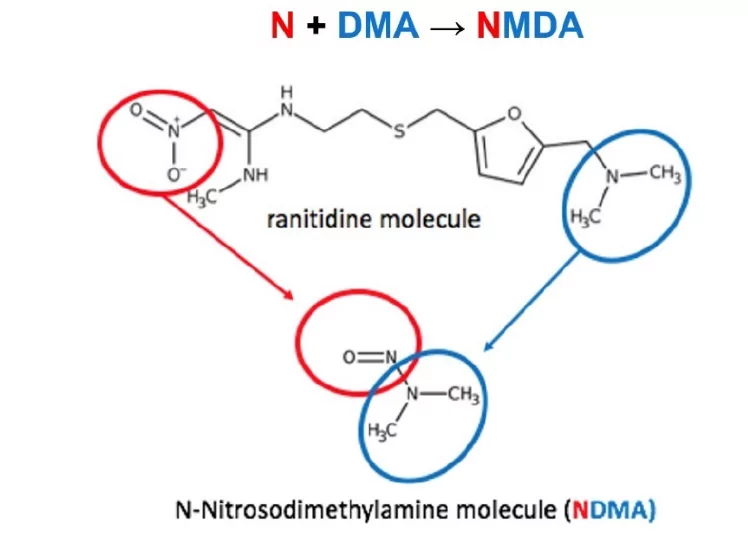
Perhaps as a consequence, this triggered the EMA to introduce a second, far more wider ranging process, Article 5(3) – Information on nitrosamines for marketing authorization holders, which required evaluation of the risk of nitrosamine impurities in any human medicinal products containing chemically synthesised active pharmaceutical ingredients.
This widened the investigation from Sartans to all synthetic molecules, with an initially extremely tight deadline of 6 months, although this was later extended until October 1st 2020 and then extended again to March 2021. Accompanied by a Q&A document, the Article 5(3) set out the following requirements:
Evaluate the possibility of the presence of nitrosamines in every medicine within 6 months:
- Prioritize evaluations, starting with medicines more likely to contain nitrosamines
- Take into account findings from CHMP’s review of sartans
- Notify authorities of outcome of risk evaluations
- Test products at risk of containing any nitrosamines
- Immediately report detection of nitrosamines to authorities
- Apply for necessary changes to marketing authorizations to address nitrosamine risk
- Complete all steps within 3 years, prioritizing high risk products
The evaluation is split into 2 steps
Step 1 – Risk evaluation: Marketing Authorization Holders (MAH) should perform risk evaluation of their medicinal products containing the chemically synthesised API. MAHs were asked to prioritize products in order to establish the sequence in which their products are to be evaluated, based on the principles defined within ICH M7 and ICH Q9.
MAHs should inform the concerned Competent Authorities when the risk evaluation is concluded. If a risk of presence of nitrosamines is identified as a result of the evaluation, the MAH should proceed to Step 2.
Step 2-Confirmatory testing: If the evaluation conducted in Step 1 identified a risk of the presence of nitrosamines, confirmatory testing should be carried out. Any needed changes to the manufacturing process that are indicated should be concluded within 3 years of the publication of the notification.
MAHs should inform the relevant authorities immediately if tests confirm the presence of a nitrosamine impurity, irrespective of the amount detected. Crucially, no specific limits are mentioned.
At the same time, Health Canada, Swiss Medic and other agencies issued similar, but not identical requests, compounding the complexity of responding to each agency. Although welcome, the announcement in March of postponement of the EMA Article 5(3) brought further challenges as not all other authorities immediately modified their timelines.
In a further twist in July 2020, the EMA issued their finalized Article 5(3) report, soon afterwards also revising their associated Questions and Answers document, for the third time. Significant within this were the following changes:

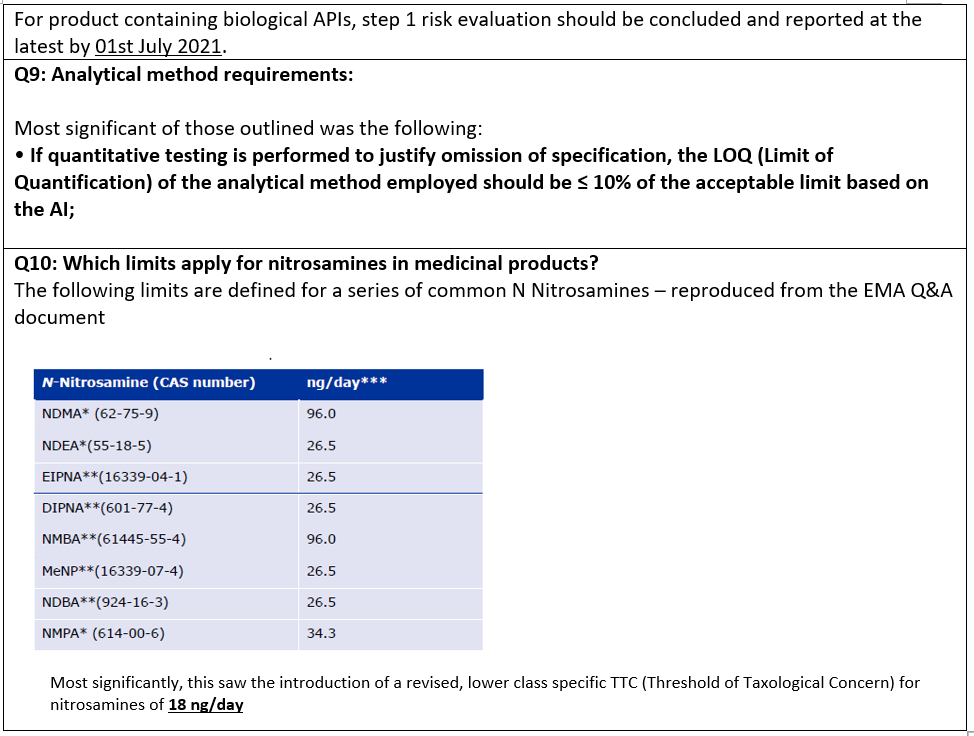
The concern is that this generic limit of 18 ng/day, when combined with a method requirement of 10% of the allowable intake (AI) for testing drug product, will render the analysis of some drug products as technically unfeasible. Will regulatory authorities continue to chase lower specifications as analytical methods leveraging high performance specialty instruments make lower detection limits possible? Will this be the end of regulatory changes? At this stage, it is impossible to say but cannot be ruled out. Such concerns will be examined further in the final blog of this series.
Related posts

The Quest for Safer, More Targeted Therapies for Patients
Our goal is to create new value through the expanded global reach and combination of Waters and Wyatt technology. The delivery of these two pieces along with our deep scientific expertise, will support our customers to make life-changing therapies more accessible.

Waters Pushes the Boundaries with Intuitive Simplicity
We believe we have created the most intuitively simple HPLC we have ever made. It is the first-ever system dedicated to the specific needs of the QC lab, and it will save our customers time, money, and stress.

Perspectives on Pharma: Change Management in the Pharmaceutical Industry
An increased emphasis on new technology and innovation for pharmaceutical companies is contributing to the supply of more consistent quality and safer products for patients.
Popular Topics
ACQUITY QDa (16) bioanalysis (11) biologics (14) biopharma (26) biopharmaceutical (36) biosimilars (11) biotherapeutics (16) case study (16) chromatography (14) data integrity (21) food analysis (12) HPLC (15) LC-MS (21) liquid chromatography (LC) (19) mass detection (15) mass spectrometry (MS) (54) method development (13) STEM (12)