L'évolution des règlements régissant les nitrosamines

En juillet 2018, l'Agence européenne des médicaments (EMA) a signalé le rappel d'un certain nombre de produits contenant l'ingrédient pharmaceutique actif (API) valsartan en raison d'une contamination par un agent cancérigène connu, la diméthyl nitrosamine, également appelée N-nitroso diméthylamine (NDMA). Cette action a déclenché une série d'événements qui ont conduit à des notifications similaires de rappels de plusieurs produits à base de sartan dans plusieurs autorités régionales. En outre, l'annonce de la découverte de la NDMA a été rapidement suivie par des rapports sur la détection d'une autre N-nitrosamine - la N-nitroso diéthylamine (NDEA).
Ces événements ont naturellement déclenché de nombreuses autorités, dont la Commission européenne (CE), pour lancer des enquêtes formelles visant à identifier les causes profondes de la contamination. La CE a ouvert une procédure le 5 juillet 2018, conformément à l'article 31 de la directive 2001/83/CE, décrivant les attentes en matière de contrôle et, si possible, d'évitement des N-Nitrosamines dans les sartans. Depuis lors, des rapports faisant état d'autres préoccupations liées à d'autres N-Nitrosamines ont continué, comme résumé ci-dessous.
Le processus initial de l'article 31 était spécifique à la classe de médicaments appelés "sartans", mais même au sein de cette classe unique, il existe un processus complexe pour établir des limites de sécurité pour chaque produit à base de sartan. Les principes définis dans le cadre de l'ICH M7 dépendent des données de sécurité relatives de chaque contaminant N-Nitrosamine et de la dose quotidienne de chaque sartan, qui peut aller de 32 mg/jour à 320 mg/jour.

De manière quelque peu controversée, ces limites ont été définies comme des limites provisoires applicables pendant une période de 2 ans, après quoi une seule limite générale de 0,03 ppm a été établie pour toute N-Nitrosamine dans tout Sartan. Il s'agit là d'une évolution importante par rapport aux limites scientifiques fondées sur la sécurité, vers une approche " aussi faible que raisonnablement possible " (ALARP), faisant des N-nitrosamines un " cas spécial ".
Dans le même temps, d'autres autorités réglementaires ont pris des mesures similaires, notamment Swiss Medic, Santé Canada et une pléthore d'autres. La FDA des États-Unis a adopté une approche quelque peu différente en demandant que la détection de toute N-nitrosamine, à quelque niveau que ce soit, soit signalée. La FDA a précisé sa pensée dans une lettre d'avis général envoyée aux détenteurs d'autorisations de mise sur le marché individuelles par le biais d'une série d'exigences spécifiques :
- L'analyse des lots pour vérifier l'absence de nitrosamine détectable dans l'IPA doit se poursuivre, à moins que le producteur d'IPA n'ait démontré que son procédé ne présente pas de risque de produire des nitrosamines détectables, conformément aux directives (par exemple, ICH Q7).
Il s'agit notamment de démontrer que :
- les matières premières, y compris les produits intermédiaires fournis par le vendeur, ne contiennent pas de nitrosamines détectables ou des quantités si faibles qu'elles peuvent être purgées de manière à ce que l'IPA ne contienne pas de quantités détectables de nitrosamines ; et
- Les matières premières utilisées dans le processus, y compris les solvants et les catalyseurs récupérés, ne contiennent pas de quantités détectables de nitrosamines.
En se concentrant également sur les matières premières, les solvants et les catalyseurs utilisés dans le processus, le champ d'investigation a été considérablement étendu. À bien des égards, cela annonçait ce qui allait suivre.
En septembre 2019, la situation relative aux N-Nitrosamines a radicalement changé. Des rapports de l'EMA, de la FDA et de la presse grand public, notamment le New York Times, ont fait état de la détection de NDMA dans la ranitidine, précisant qu'en raison de sa structure unique, elle pouvait se dégrader pour générer de la NDMA à partir de la molécule elle-même.
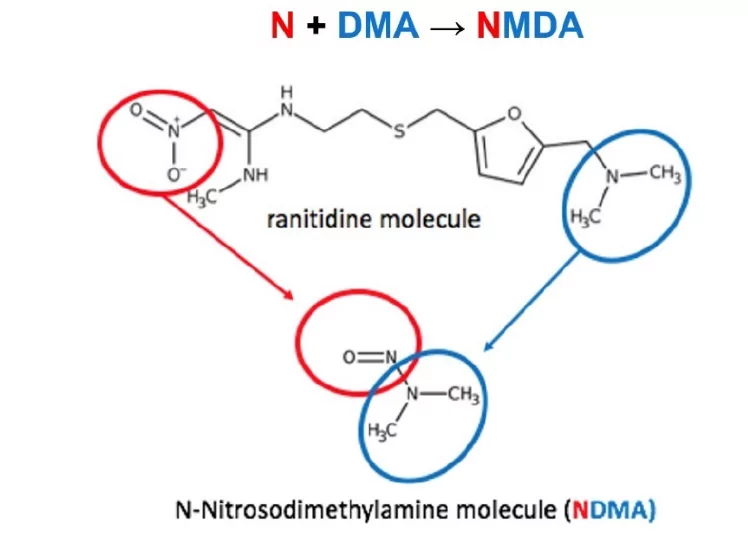
Peut-être en conséquence, cela a incité l'EMA à introduire un deuxième processus, beaucoup plus large, l'article 5(3) - Information sur les nitrosamines pour les détenteurs d'autorisation de mise sur le marché, qui exigeait l'évaluation du risque d'impuretés de nitrosamines dans tout médicament humain contenant des ingrédients pharmaceutiques actifs synthétisés chimiquement.
Cela a permis d'élargir l'enquête sur les sartans à toutes les molécules synthétiques, avec un délai initialement extrêmement serré de 6 mois, bien que celui-ci ait ensuite été prolongé jusqu'au1er octobre 2020, puis à nouveau jusqu'en mars 2021. Accompagné d'un document de questions-réponses, l'article 5, paragraphe 3, énonce les exigences suivantes :
Évaluez la possibilité de la présence de nitrosamines dans chaque médicament dans les 6 mois :
- Prioriser les évaluations, en commençant par les médicaments les plus susceptibles de contenir des nitrosamines.
- Tenir compte des conclusions de l'examen des sartans par le CHMP.
- Informer les autorités des résultats des évaluations des risques
- Testez les produits susceptibles de contenir des nitrosamines.
- Signalez immédiatement la détection de nitrosamines aux autorités.
- Demander les modifications nécessaires aux autorisations de mise sur le marché pour faire face au risque lié aux nitrosamines.
- Réaliser toutes les étapes dans un délai de 3 ans, en donnant la priorité aux produits à haut risque.
L'évaluation est divisée en 2 étapes
Étape 1 - Évaluation des risques : Les détenteurs d'autorisation de mise sur le marché (TAMM) doivent effectuer une évaluation des risques de leurs médicaments contenant l'IPA synthétisé chimiquement. Les TAMM ont été invités à classer les produits par ordre de priorité afin d'établir la séquence dans laquelle leurs produits doivent être évalués, sur la base des principes définis dans ICH M7 et ICH Q9.
Les TAMM doivent informer les autorités compétentes concernées lorsque l'évaluation des risques est terminée. Si un risque de présence de nitrosamines est identifié à la suite de l'évaluation, le TAMM devrait passer à l'étape 2.
Étape 2 - Essais de confirmation : Si l'évaluation menée à l'étape 1 a identifié un risque de présence de nitrosamines, des tests de confirmation doivent être effectués. Toute modification nécessaire du procédé de fabrication qui est indiquée doit être conclue dans les 3 ans suivant la publication de la notification.
Les TAMM doivent informer immédiatement les autorités compétentes si les tests confirment la présence d'une impureté de type nitrosamine, quelle que soit la quantité détectée. Il est important de noter qu'aucune limite spécifique n'est mentionnée.
Dans le même temps, Santé Canada, Swiss Medic et d'autres agences ont émis des demandes similaires, mais pas identiques, ce qui rend encore plus complexe la réponse à chaque agence. Bien que bienvenue, l'annonce en mars du report de l'article 5(3) de l'EMA a posé de nouveaux défis, car toutes les autres autorités n'ont pas immédiatement modifié leurs délais.
En juillet 2020, l'EMA a publié son rapport final sur l'article 5(3) et, peu après, a également révisé pour la troisième fois son document de questions et réponses associé. Les changements les plus importants sont les suivants :

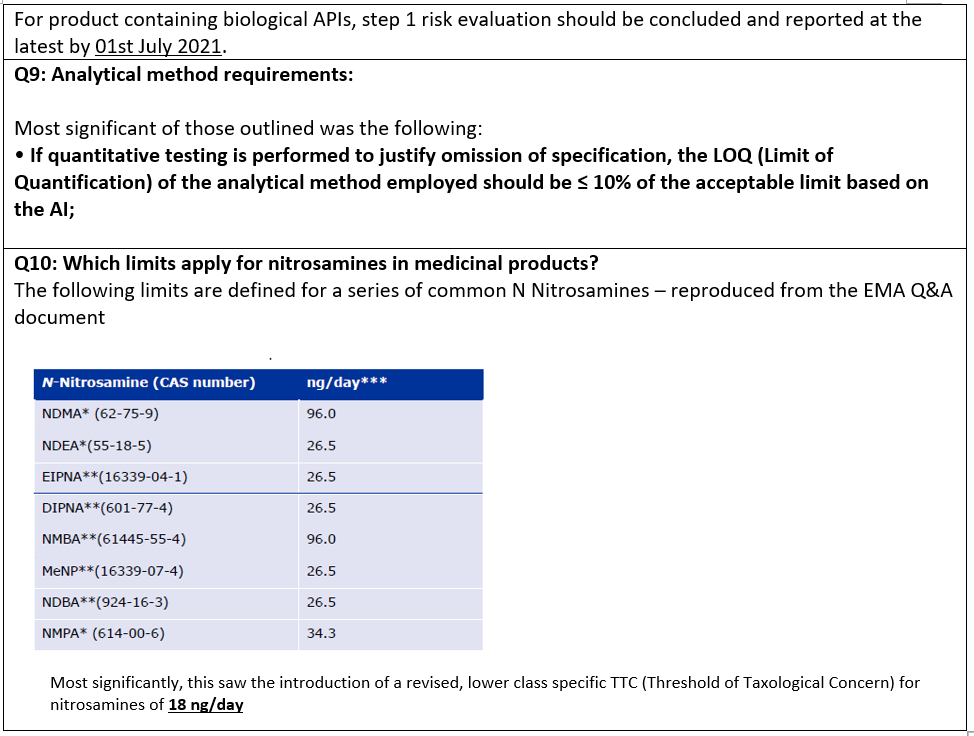
La préoccupation est que cette limite générique de 18 ng/jour, lorsqu'elle est combinée à une exigence de méthode de 10% de la dose admissible (AI) pour tester le produit pharmaceutique, rendra l'analyse de certains produits pharmaceutiques techniquement irréalisable. Les autorités réglementaires continueront-elles à rechercher des spécifications plus basses, car les méthodes analytiques utilisant des instruments spécialisés de haute performance permettent d'abaisser les limites de détection ? Sera-ce la fin des changements réglementaires ? À ce stade, il est impossible de le dire, mais on ne peut l'exclure. Ces préoccupations seront examinées plus en détail dans le dernier blog de cette série.
Postes connexes

La quête de thérapies plus sûres et plus ciblées pour les patients
Notre objectif est de créer une nouvelle valeur grâce à l'extension de la portée mondiale et à la combinaison des technologies de Waters et de Wyatt. L'apport de ces deux éléments, ainsi que notre profonde expertise scientifique, aidera nos clients à rendre plus accessibles les thérapies qui changent la vie.

Waters repousse les limites avec une simplicité intuitive
Nous pensons avoir créé le système HPLC le plus intuitif et le plus simple que nous ayons jamais fabriqué. Il s'agit du tout premier système dédié aux besoins spécifiques du laboratoire de contrôle qualité, et il permettra à nos clients d'économiser du temps, de l'argent et du stress.

Perspectives on Pharma : Gestion du changement dans l'industrie pharmaceutique
L'importance accrue accordée aux nouvelles technologies et à l'innovation par les entreprises pharmaceutiques contribue à la fourniture de produits de qualité plus constante et plus sûre pour les patients.
Sujets populaires
ACQUITY QDa (16) bioanalyse (11) produits biologiques (14) biopharma (26) biopharmaceutique (36) biosimilaires (11) biothérapeutique (16) étude de cas (16) chromatographie (14) intégrité des données (21) analyse alimentaire (12) HPLC (15) LC-MS (21) chromatographie liquide (LC) (19) détection de masse (15) spectrométrie de masse (MS) (54) développement de méthodes (13) STEM (12)