Faire progresser la recherche omique et réduire les biais grâce à une nouvelle technologie MS pour les expériences d'acquisition indépendante des données

Les tests ciblés basés sur la LC-MS sont de plus en plus appliqués dans le domaine Omics post-découverte, en mettant l'accent sur la validation, la première des nombreuses phases de la recherche translationnelle, et dans les études visant à mieux comprendre les systèmes biologiques en termes de développement et de traitement des médicaments.
La compréhension du contexte des résultats analytiques est le moteur de la recherche actuelle, et donc du développement de méthodes d'acquisition LC-MS qui peuvent fournir des informations qualitatives et quantitatives en une seule expérience.
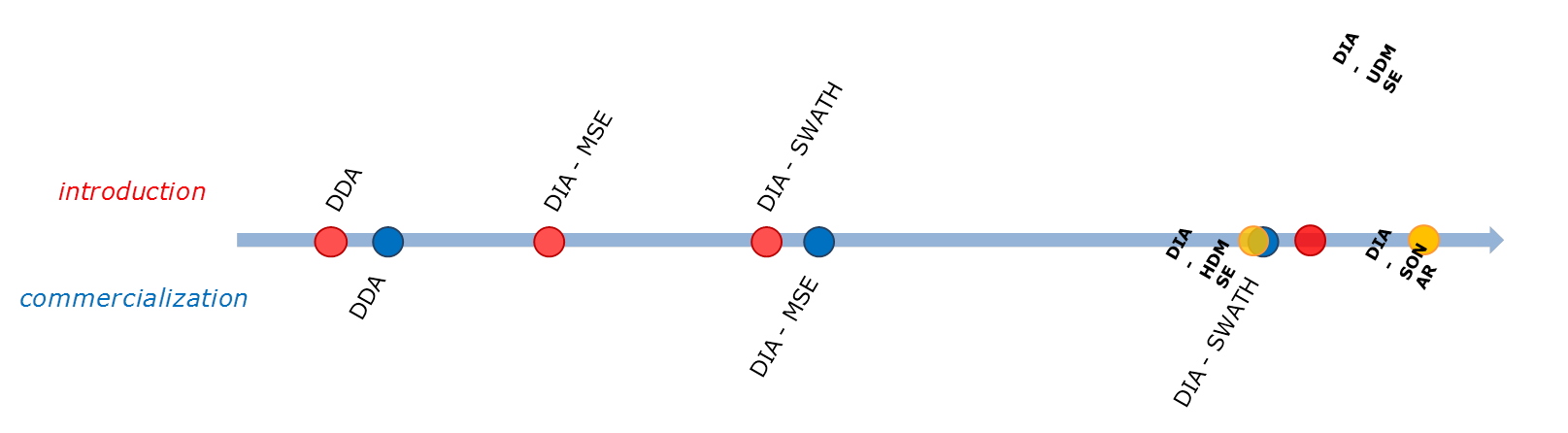
L'acquisition indépendante des données (DIA) est une technique essentielle pour les laboratoires qui doivent identifier qualitativement des métabolites, des lipides et des protéines dans des échantillons complexes et extraire des informations quantitatives à haute résolution.
Dans une expérience DIA, le comportement du spectromètre de masse ne change pas en fonction des données générées. Cela signifie que les résultats ne sont pas biaisés et qu'aucune donnée n'est perdue par le système de spectrométrie de masse qui passe d'un mode d'acquisition à un autre ou à des limites de seuil expérimentales, ce qui est une préoccupation pour les approches d'acquisition dépendantes des données (DDA).
Un nouveau mode d'acquisition, le MS/MS 2D, a été présenté pour la première fois par Richardson, et al, à l'ASMS en 2015, et a été récemment introduit commercialement sous le nom de SONAR de Waters.
"Avec la maturation de la recherche en protéomique, les scientifiques recueillent déjà la majorité des informations disponibles à partir des protéines. Ce qu'ils veulent, c'est pouvoir aborder une protéine ou des peptides spécifiques avec une hypothèse, et utiliser la quantification MS/MS ciblée pour étudier leurs idées sans mettre en place des méthodes ou des expériences supplémentaires.
"Désormais, avec l'acquisition de données SONAR, ils peuvent effectuer une analyse tout-en-un plus sélective. Compatible avec les séparations UPLC à haute vitesse, c'est un flux de travail plus efficace pour une analyse qualitative et quantitative plus précise en une seule injection. "(tiré de World HUPO 2016)
En mode SONAR, le quadripôle est rapidement balayé sur la plage m/z d'intérêt, en alternant les balayages de basse énergie et de haute énergie et en recueillant des données qualitatives et quantitatives pour tous les précurseurs et tous les produits. L'électronique de balayage rapide échantillonne 200 positions du quadripôle par balayage avec des temps de balayage typiques de 0,1 à 0,5 seconde (pour chaque niveau d'énergie). Le quadripôle peut balayer à >10 000 amu/sec, >2000 spectres/sec et est compatible avec les séparations chromatographiques et électrophorétiques à grande vitesse.
Permettant aux chercheurs d'effectuer une manipulation avancée des données, les données SONAR sont compatibles avec les flux de travail analytiques qui utilisent les logiciels Waters Progenesis QI et Symphony, ainsi qu'avec des logiciels tiers tels que Skyline. SONAR est disponible sur le spectromètre de masse Xevo G2-XS QTof de Waters sous le contrôle du logiciel MassLynx.
Cette méthode offre des avantages en fournissant des données MS/MS quantitatives à des vitesses UPLC à partir d'une méthode d'acquisition indépendante des données (DIA) pour une utilisation dans des échantillons très complexes. Un poster récent intitulé Advances in Targeted Omics Quantitation Using a Novel Scanning Quadrupole DIA Method fournit de plus amples informations.
Dans un récent webinaire intitulé Qualitative and Quantitative Proteomics - Novel DIA and Processing Strategies for Complex Profiling, publié dans le Journal of Proteomics, le Dr James Langridge de Waters explique le fonctionnement de SONAR, et le Dr Paul Skipp, professeur associé/directeur de centre, Centre for Proteomic Research de l'Université de Southampton, explique comment il permet le profilage global et la quantification ciblée du protéome de la peau humaine.
Plus de ressources :
- SONAR pour les applications omiques
- Poster : Progrès dans la quantification des omiques ciblés à l'aide d'une nouvelle méthode de DIA quadripolaire à balayage
- Webinaire à la demande : Protéomique qualitative et quantitative - Nouvelles stratégies de DIA et de traitement pour un profilage complexe
Vous faites du DIA #massspec pour de la #protéomique ciblée ? Découvrez SONAR : Arrêtez de regarder, commencez à voir. Vidéo : https://t.co/IdKb2znQNp
- Waters Corporation (@WatersCorp) 20 septembre 2016
Postes connexes

L'avenir du dépistage du cancer du sein ?
Chez Waters, nous innovons constamment nos systèmes de spectrométrie de masse pour améliorer la santé humaine, comme cette avancée révolutionnaire dans la détection du cancer du sein.

Les flacons d'échantillon à travers les âges
Depuis les années 1700 jusqu'à l'ère moderne, les flacons d'échantillons font partie intégrante du flux de travail des laboratoires d'analyse. Aujourd'hui encore, nous testons et certifions l'ensemble du kit de flacons.

Comment une meilleure sélectivité dans l'analyse des immunosuppresseurs peut aider les patients transplantés d'organes
Les jeux de calibrateurs, de contrôles et de standards internes Waters MassTrak Immunosuppressant, bientôt disponibles, sont développés pour aider les laboratoires à utiliser le LC-MS/MS en toute confiance dans leurs résultats de dosage.
Sujets populaires
ACQUITY QDa (16) bioanalyse (11) produits biologiques (14) biopharma (26) biopharmaceutique (36) biosimilaires (11) biothérapeutique (16) étude de cas (16) chromatographie (14) intégrité des données (21) analyse alimentaire (12) HPLC (15) LC-MS (21) chromatographie liquide (LC) (19) détection de masse (15) spectrométrie de masse (MS) (54) développement de méthodes (13) STEM (12)