La evolución de la normativa sobre nitrosaminas

En julio de 2018, la Agencia Europea de Medicamentos (EMA) notificó la retirada de varios productos que contenían el ingrediente farmacéutico activo (API) valsartán debido a la contaminación con un carcinógeno conocido, la dimetil nitrosamina, también conocida como N-nitroso dimetilamina (NDMA). Esta acción desencadenó una cadena de acontecimientos que condujo a notificaciones similares de retirada de varios productos de sartán en múltiples autoridades regionales. Además, al anuncio del descubrimiento de la NDMA le siguieron rápidamente informes sobre la detección de otra N-Nitrosamina: la N-Nitroso dietilamina (NDEA).
Estos acontecimientos, comprensiblemente, desencadenaron que muchas autoridades, incluida la Comisión Europea (CE), iniciaran investigaciones formales destinadas a identificar las causas fundamentales de la contaminación. La CE inició un procedimiento el 5 de julio de 2018, de conformidad con el artículo 31 de la Directiva 2001/83/CE. en el que se exponen las expectativas de control y, cuando sea posible, de evitación de las N Nitrosaminas en los sartanes. Desde entonces, han continuado los informes de nuevas preocupaciones relacionadas con otras N-Nitrosaminas, como se resume a continuación.
El proceso inicial del artículo 31 era específico para la clase de medicamentos denominados "sartanes", pero incluso dentro de esta única clase existe un proceso complejo para establecer los límites de seguridad de cada producto basado en sartán. Los principios definidos en la ICH M7 dependen de los datos de seguridad relativos de cada contaminante N-Nitrosamina y de la dosis diaria de cada sartán, que puede oscilar entre 32 mg/día y 320 mg/día.

De forma algo controvertida, se definieron como límites provisionales aplicables durante un periodo de dos años, tras el cual se estableció un único límite general de 0,03 ppm para cualquier N-Nitrosamina en cualquier sartán. Esto representa un cambio significativo de los límites científicos basados en la seguridad, hacia un enfoque "tan bajo como sea razonablemente práctico" (ALARP), haciendo de las N-Nitrosaminas un "caso especial".
Al mismo tiempo, otras autoridades reguladoras, como Swiss Medic, Health Canada y otras, adoptaron una medida similar. La FDA de los Estados Unidos adoptó un enfoque algo diferente, solicitando que se notificara la detección de cualquier N-Nitrosamina a cualquier nivel. La FDA reveló además su pensamiento en una carta de asesoramiento general enviada a los titulares de autorizaciones de comercialización individuales a través de una serie de requisitos específicos:
- Las pruebas por lotes para verificar que no hay nitrosaminas detectables en el API deben continuar, a menos que el productor del API haya demostrado que su proceso no corre el riesgo de producir nitrosaminas detectables de acuerdo con las directrices (por ejemplo, ICH Q7).
Esto incluye demostrar que:
- los materiales de partida, incluidos los productos intermedios suministrados por el proveedor, no tienen nitrosaminas detectables o se pueden purgar cantidades tan bajas que garanticen que el API no contiene cantidades detectables de nitrosaminas, y
- Las materias primas utilizadas en el proceso, incluidos los disolventes y catalizadores recuperados, no contienen cantidades detectables de nitrosaminas.
Al centrarse también en los materiales de partida, los disolventes y los catalizadores utilizados en el proceso, el alcance de las investigaciones se amplió considerablemente. Esto fue, en muchos sentidos, un presagio de lo que siguió.
En septiembre de 2019 la situación relativa a las N-Nitrosaminas cambió radicalmente. Los informes de la EMA, la FDA y la prensa general, incluido el New York Times, informaron de la detección de NDMA en la ranitidina, afirmando que, debido a su estructura única, podría degradarse para generar NDMA a partir de la propia molécula.
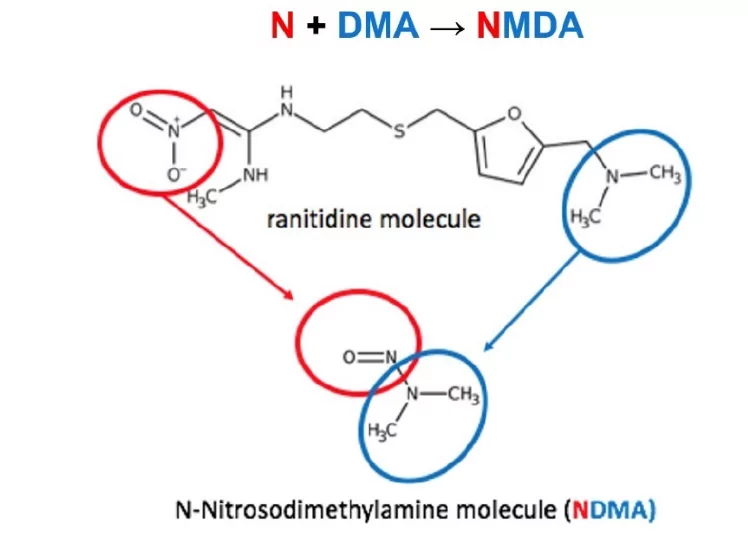
Tal vez como consecuencia, esto provocó que la EMA introdujera un segundo proceso de mucho mayor alcance, el apartado 3 del artículo 5 - Información sobre las nitrosaminas para los titulares de autorizaciones de comercialización, que exigía la evaluación del riesgo de impurezas de nitrosaminas en cualquier medicamento de uso humano que contuviera ingredientes farmacéuticos activos de síntesis química.
Esto amplió la investigación de los sartanes a todas las moléculas sintéticas, con un plazo inicialmente muy ajustado de 6 meses, aunque posteriormente se amplió hastael 1 de octubre de 2020 y luego se volvió a ampliar hasta marzo de 2021. Acompañado de un documento de preguntas y respuestas, el apartado 3 del artículo 5 establecía los siguientes requisitos:
Evaluar la posibilidad de la presencia de nitrosaminas en cada medicamento en un plazo de 6 meses:
- Priorizar las evaluaciones, empezando por los medicamentos con mayor probabilidad de contener nitrosaminas
- Tener en cuenta los resultados de la revisión del CHMP sobre los sartanes
- Notificar a las autoridades el resultado de las evaluaciones de riesgo
- Analizar los productos con riesgo de contener nitrosaminas
- Informar inmediatamente a las autoridades de la detección de nitrosaminas
- Solicitar los cambios necesarios en las autorizaciones de comercialización para abordar el riesgo de las nitrosaminas
- Completar todos los pasos en un plazo de 3 años, dando prioridad a los productos de alto riesgo
La evaluación se divide en dos etapas
Paso 1 - Evaluación del riesgo: Los titulares de la autorización de comercialización (TAC) deben realizar la evaluación de riesgos de sus medicamentos que contienen el API de síntesis química. Se pidió a los titulares de la autorización de comercialización que priorizaran los productos para establecer la secuencia de evaluación de sus productos, basándose en los principios definidos en ICH M7 e ICH Q9.
El titular de la autorización de comercialización deberá informar a las Autoridades Competentes correspondientes una vez concluida la evaluación del riesgo. Si se identifica un riesgo de presencia de nitrosaminas como resultado de la evaluación, el titular de la autorización de comercialización deberá pasar a la etapa 2.
Etapa 2 - Pruebas de confirmación: Si la evaluación realizada en la Etapa 1 identificó un riesgo de presencia de nitrosaminas, deben realizarse pruebas de confirmación. Cualquier cambio necesario en el proceso de fabricación que se indique deberá concluirse en un plazo de 3 años a partir de la publicación de la notificación.
Los HMA deben informar inmediatamente a las autoridades competentes si las pruebas confirman la presencia de una impureza de nitrosamina, independientemente de la cantidad detectada. Lo más importante es que no se mencionan límites específicos.
Al mismo tiempo, Health Canada, Swiss Medic y otras agencias emitieron solicitudes similares, pero no idénticas, lo que complicó la respuesta a cada agencia. Aunque se acogió con satisfacción, el anuncio en marzo del aplazamiento del apartado 3 del artículo 5 de la EMA supuso nuevos retos, ya que no todas las demás autoridades modificaron inmediatamente sus plazos.
En un nuevo giro, en julio de 2020, la EMA publicó su informe final del artículo 5(3), y poco después también revisó su documento asociado de preguntas y respuestas, por tercera vez. Los cambios más significativos fueron los siguientes:

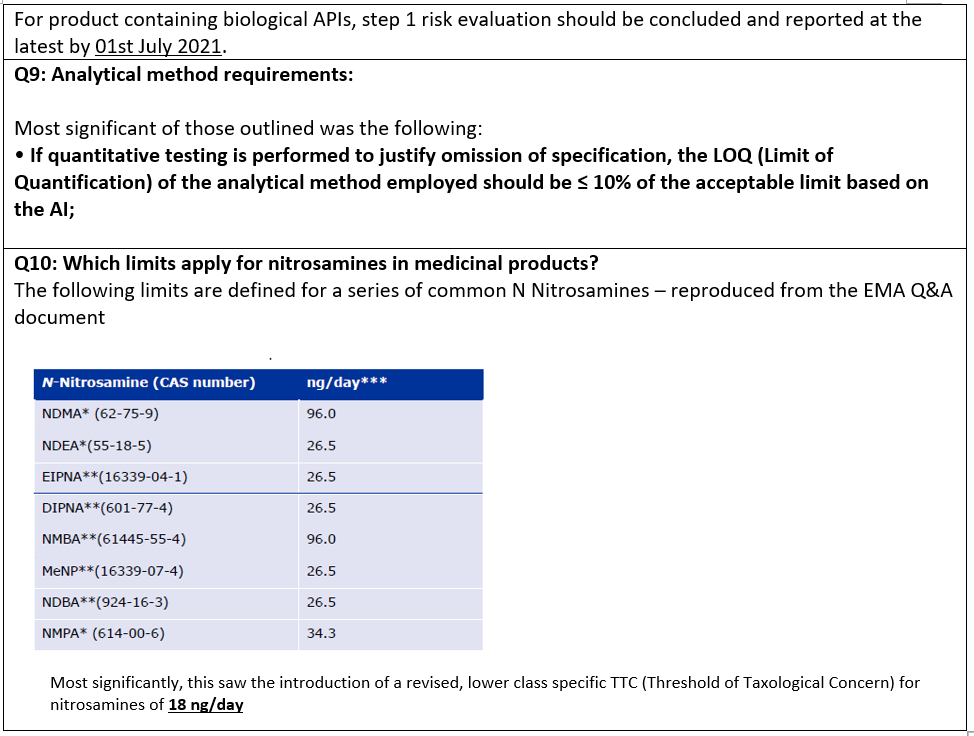
La preocupación es que este límite genérico de 18 ng/día, cuando se combina con un requisito de método del 10% de la ingesta permitida (AI) para probar el producto farmacéutico, hará que el análisis de algunos productos farmacéuticos sea técnicamente inviable. ¿Continuarán las autoridades reguladoras persiguiendo especificaciones más bajas a medida que los métodos analíticos que aprovechan los instrumentos especializados de alto rendimiento hacen posible límites de detección más bajos? ¿Será este el fin de los cambios normativos? En este momento, es imposible decirlo, pero no se puede descartar. En el último blog de esta serie se examinarán más a fondo estas cuestiones.
Puestos relacionados

La búsqueda de terapias más seguras y específicas para los pacientes
Nuestro objetivo es crear nuevo valor mediante la ampliación del alcance mundial y la combinación de la tecnología de Waters y Wyatt. La entrega de estas dos piezas, junto con nuestra profunda experiencia científica, ayudará a nuestros clientes a hacer más accesibles las terapias que cambian vidas".

Waters supera los límites con sencillez intuitiva
Creemos que hemos creado el HPLC más intuitivamente sencillo que jamás hayamos fabricado. Es el primer sistema dedicado a las necesidades específicas del laboratorio de control de calidad, y ahorrará a nuestros clientes tiempo, dinero y estrés.

Perspectives on Pharma: Gestión del cambio en la industria farmacéutica
El mayor énfasis de las empresas farmacéuticas en las nuevas tecnologías y la innovación está contribuyendo al suministro de productos de calidad más constante y seguros para los pacientes.
Temas populares
ACQUITY QDa (16) bioanálisis (11) biológicos (14) biofarmacia (26) biofarmacéutica (36 ) biosimilares (11) bioterapéutica (16) estudio de caso (16) cromatografía (14) integridad de los datos (21) análisis de alimentos (12) HPLC (15) LC-MS (21 ) cromatografía líquida (LC) (19) detección de masas (15) espectrometría de masas (EM) (54 ) desarrollo de métodos (13) STEM (12)