Data Integrity Matters | Unjustified Invalidation of Orphan Data

Orphan data: To delete or not to delete
In February 2017 the U.S. FDA issued a brief Warning Letter on one single topic: Unjustified invalidation (and deletion) of orphan data.
“Our investigators reviewed audit trails from various stand-alone pieces of laboratory equipment you used to perform high performance liquid chromatography (HPLC) and gas chromatography (GC) analyses. Our investigators found that you had deleted entire chromatographic sequences and individual injections from your stand-alone computers. Without providing scientific justification, you repeated analyses until you obtained acceptable results. You failed to investigate original out-of-specification or otherwise undesirable test results, and you only documented passing test results in logbooks and preparation notebooks. You relied on these manipulated test results and incomplete records to support batch release decisions.”
Are we even asking the right the question?
 Orphan data may exist for any number of scientific or operational reasons, but in today’s environment – until the regulators trust the companies again – each piece of orphan data is suspect and may contain evidence of data tampering.
Orphan data may exist for any number of scientific or operational reasons, but in today’s environment – until the regulators trust the companies again – each piece of orphan data is suspect and may contain evidence of data tampering.
Chromatographic analysis may not be an exact science, but it relies a great deal on the robustness of the original method (including its regular review and update) and the consistent performance of a large number of individual components: Sample, standard and solvent preparation steps, pumps, injectors, detectors and data capture hardware, laboratory temperature, column robustness and performance, user skills and training, degree of automation in the process, the opportunities for manual errors in data entry or in calculated final results, the clarity of SOPs, traceability (or chain of custody) of the test preparations, skill – and knowledge of the reviewers.
Failure or misstep in any of these contributing attributes may easily end up in a set of results that need to be invalidated and repeated. The critical question that must be asked is:
“If the analysts or lab staff were trying to invalidate ‘undesirable’ results, would the review process distinguish this egregious behavior from expected normal error correction?”
The easiest and most important step is to never delete orphan data
Any reviewer or QA professional then has the opportunity to review the invalidated data and judge if its invalidation is scientifically justified. But equally, it is key not to attempt a switch from a state of 0% review of earlier created data and associated audit trails, to an expectation of 100% review of all data and all meta data including all audit trails. Aside from the productivity concerns of reviewing every action of every analyst before accepting a result as true, the skills and knowledge of the reviewers in the topic they are reviewing may need to be substantially assessed and enhanced.
But how do we manage orphan data results post-review? Should all the data follow the results forever?
For this, I look at how the World Health Organization (WHO) defined a Summary Report.
“Once a detailed review of all the electronic data is completed, and a Summary Report is created and reviewed which accurately reflects all the data, then the summary may be used for further decision making.”
Providing some expert reviewer has assessed the summary or final data set or “report” and is confident that any orphan data or results created can scientifically be invalidated and ignored, that the data matches the data found in the electronic records, and that the “final” values reported in the summary are to be trusted – from this point on the information in the summary can be viewed as “official” and could be leveraged in any further reporting requirements or decision making, i.e., in study reports or in Certificates of Analysis.
In a similar fashion to the evolution of Computer System Validation (CSV) practices, it will take time for industry and regulators to agree on how to successfully apply a risk-based approach to “review of complete data.” Key to this challenge is defining which data needs to be reviewed, in what format, how often, and how to document a review process (outside of key stoke monitoring or videoing the entire analytical analysis process).
This applies to all kinds of testing and recording of data, but the reliance on analyst’s knowledge and skill to correctly interpret chromatographic data makes this a key area for defining a robust review process versus implementing impossible controls.
Infographic: Meeting the technical requirements for compliance of laboratory computerized systems
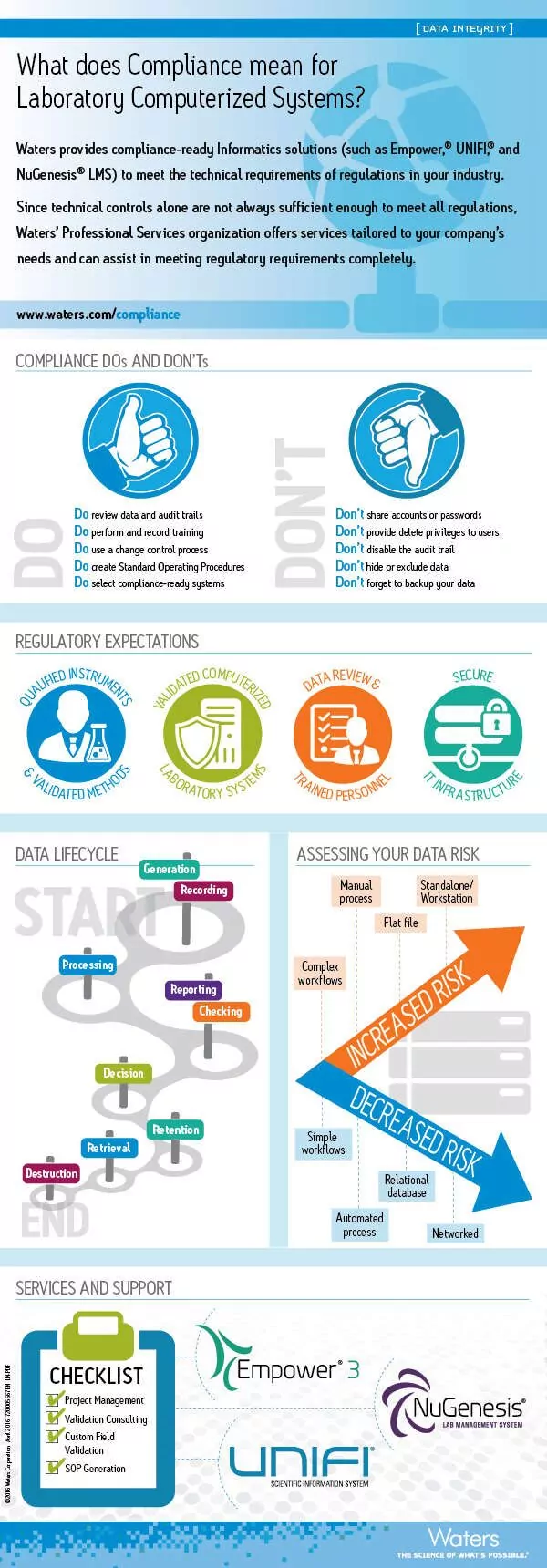
Read more articles in Heather Longden’s blog series, Data Integrity Matters.
More resources:
- On-demand webinar: Why is Electronic CDS Data a Major Data Integrity Concern for Regulators?
- What Does Compliance Mean for Laboratory Computerized Systems? Read our white paper
- Learn more about ensuring Quality through compliance with Waters
Related posts

Fostering a Supportive Community for Veterans
I am honored and privileged to be part of the community of veterans at Waters that fosters an opportunity to connect, share our experiences and support one another.

The Jim Waters Society Inductees Reflect Inventive Achievement
Four new Jim Waters Society inductees represent Waters’ top scientists and engineers for remarkable achievements.

Veterans Bring Leadership to the Waters Team
At Waters, we strive to unite and celebrate our diverse employee population. To this end, we have established Employee Circles, which create safe spaces where employees can share their experiences and learn from each other. Just before the pandemic, we launched the Veterans Circle—an employee resource group designed for and composed of Waters veterans and…
Popular Topics
ACQUITY QDa (16) bioanalysis (11) biologics (14) biopharma (26) biopharmaceutical (36) biosimilars (11) biotherapeutics (16) case study (16) chromatography (14) data integrity (21) food analysis (12) HPLC (15) LC-MS (21) liquid chromatography (LC) (19) mass detection (15) mass spectrometry (MS) (54) method development (13) STEM (12)